Singapore Med J 2017; 58(5): 281 doi: 10.11622/smedj.2017040
Anaemia with Laugier-Hunziker Syndrome: a diagnostic dilemma
Muhammad Bilal Abid1, Paras Mughal2, Muhammad Abbas Abid3
Author Information >Copyright and License information >
1Department of Haematology-Oncology, National University Cancer Institute, Singapore 2Department of Orthodontics, Mount Elizabeth Hospital, Singapore 3Department of Otorhinolaryngology-Head and Neck Surgery, John Hopkins School of Medicine, Baltimore, USA
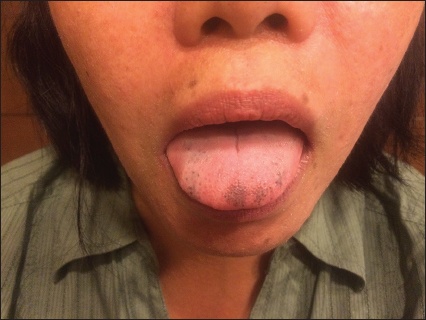
We herein describe the case of a 39-year-old woman with no significant medical history, who presented with a two-week history of painless, non-pruritic, macular hyperpigmentation of the tongue (Fig. 1). She also had brown-coloured pigmentation on the tip of all fingers and toes with pigmented vertical bands on the thumbnails. There was no koilonychia or darkening of palmar creases. The patient was found to have profound hypochromic, microcytic anaemia (haemoglobin 4.4 g/dL) on blood investigations even though she was asymptomatic upon presentation. She reported no drug consumption, systemic symptoms or gastrointestinal (GI) red flags such as loss of appetite; weight loss; night sweats; dysphagia; new-onset or progressively worsening heartburn or abdominal pain; change in colour, consistency or calibre of bowel movements; and melaena or haematochezia. Her diet included adequate haematinics and she had no family or personal history of anaemia, skin conditions or intestinal polyposis.
Fig. 1
Photograph shows macular hyperpigmentation of the tongue of a 39-year-old woman who was diagnosed with Laugier-Hunziker Syndrome.
Investigations showed elevated red cell distribution width, mild thrombocytosis and a Mentzer index > 13. Iron studies further confirmed a diagnosis of iron deficiency anaemia attributable to menstrual blood loss. She had had menorrhagia over the preceding eight months, with heavier menstrual flow during the first two days of her monthly cycle and each cycle lasting 5–6 days. Normal haemodynamic parameters and normal plasma electrolyte, cortisol and adrenocorticotropic hormone levels ruled out Addison’s disease, while a lack of family history, GI symptoms and negative findings in endoscopy and colonoscopy ruled out Peutz-Jeghers Syndrome (PJS). Small bowel endoscopy was not performed. The tongue and skin pigmentation was diagnosed as Laugier-Hunziker Syndrome (LHS) based on the patient’s clinical presentation and the histopathologic appearance of an increase in melanin in the basal layer of epidermis. The patient was treated with iron supplements, reassured about her skin condition and discharged with a gynaecology follow-up visit.
The concurrent presentation of two discrete disorders with overlapping features is a common occurrence in clinical medicine. In general, the abrupt appearance of macular, mucocutaneous pigmentation may be considered alarming and is likely to be associated with a variety of syndromes, especially when it is concurrent with profound anaemia. LHS is an acquired disorder characterised by diffuse hyperpigmentation of the oral mucosa and longitudinal melanonychia,(1,2) whereas PJS is an autosomal dominant disease characterised by hamartomatous polyps in the GI tract and may also have oral or mucocutaneous melanosis.(3,4) Diagnosis may be further convoluted in a resource-limited healthcare setting. The importance of a comprehensive history and a thorough physical examination cannot be adequately emphasised, especially when confronted with such clinical dilemmas.
LHS is rare, with nearly 180 cases reported thus far, and has a 2:1 female predilection. It is considered to be a benign disease with no systemic manifestation or malignant potential;(5) important differential diagnoses include PJS and Addison’s disease, among other causes of oral and acral pigmentation such as drugs and heavy metals.(6,7) Overlapping clinical features may pose diagnostic problems in patients with concurrent cutaneous and systemic manifestations. Given the limited healthcare resources in the Asian continent and the fact that there could be overlaps between manifestations of any two clinical conditions, meticulous attention to history and physical examination is needed in differentiating problems arising from two separate pathologies rather than a syndromic presentation.
References Laugier P, Hunziker N. [Essential lenticular melanic pigmentation of the lip and cheek mucosa].Arch Belg Dermatol Syphiligr. 1970; 26:391-9. French. Gerbig AW, Hunziker T. Idiopathic lenticular mucocutaneous pigmentation or Laugier-Hunziker syndrome with atypical features.Arch Dermatol. 1996; 132:844-5. Chang CJ, Nelson JS. Q-switched ruby laser treatment of mucocutaneous melanosis associated with Peutz-Jeghers syndrome.Ann Plast Surg. 1996; 36:394-7. Bartholomew LG, Dahlin DC, Waugh JM. Intestinal polyposis associated with mucocutaneous melanin pigmentation Peutz-Jeghers syndrome; review of literature and report of six cases with special reference to pathologic findings.Gastroenterology. 1957; 32:434-51. Kemmett D, Ellis J, Spencer MJ, Hunter JA. The Laugier-Hunziker syndrome--a clinical review of six cases.Clin Exp Dermatol. 1990; 15:111-4. Lampe AK, Hampton PJ, Woodford-Richens K, et al. Laugier-Hunziker syndrome: an important differential diagnosis for Peutz–Jeghers syndrome.J Med Genet. 2003; 40:e77. Eisen D. Disorders of pigmentation in the oral cavity.Clin Dermatol. 2000; 18:579-87.
REFERENCES
1. Laugier P, Hunziker N. [Essential lenticular melanic pigmentation of the lip and cheek mucosa]. Arch Belg Dermatol Syphiligr 1970; 26:391-9. French.
PMid:5515564
2. Gerbig AW, Hunziker T. Idiopathic lenticular mucocutaneous pigmentation or Laugier-Hunziker syndrome with atypical features. Arch Dermatol 1996; 132:844-5. https://doi.org/10.1001/archderm.1996.03890310136032
3. Chang CJ, Nelson JS. Q-switched ruby laser treatment of mucocutaneous melanosis associated with Peutz-Jeghers syndrome. Ann Plast Surg 1996; 36:394-7. https://doi.org/10.1097/00000637-199604000-00012
4. Bartholomew LG, Dahlin DC, Waugh JM. Intestinal polyposis associated with mucocutaneous melanin pigmentation Peutz-Jeghers syndrome; review of literature and report of six cases with special reference to pathologic findings. Gastroenterology 1957; 32:434-51.
PMid:13415265
5. Kemmett D, Ellis J, Spencer MJ, Hunter JA. The Laugier-Hunziker syndrome--a clinical review of six cases. Clin Exp Dermatol 1990; 15:111-4. https://doi.org/10.1111/j.1365-2230.1990.tb02044.x
PMid:2347100
6. Lampe AK, Hampton PJ, Woodford-Richens K, et al. Laugier-Hunziker syndrome: an important differential diagnosis for Peutz–Jeghers syndrome. J Med Genet 2003; 40:e77. https://doi.org/10.1136/jmg.40.6.e77
PMid:12807976 PMCid:PMC1735508