

Abstract
Central pontine myelinolysis (CPM) is classically described as a demyelinating condition that results from the rapid correction of hyponatraemia. CPM has also been reported to arise from hyperglycaemia in association with concomitant acidosis, hypernatraemia and hyperosmolar syndrome. Herein, we report a rare presentation of CPM, which was purely secondary to hyperosmolar hyperglycaemia. The patient presented with ataxia and pseudobulbar affect, which evolved subacutely over a duration of two weeks. It is important to note that, in addition to acute changes in osmolality, a subacute shift secondary to hyperglycaemia may also lead to CPM.
INTRODUCTION
Central pontine myelinolysis (CPM) is a noninflammatory, demyelinating condition that has been described mostly in the context of rapid correction of hyponatraemia.(1) Classical clinical manifestations range from a depressed level of consciousness to dysarthria and flaccid quadriplegia.(1,2) Recently, there has been an increase in the number of reports of CPM occurring in association with other hyperosmolar states, and with radiological lesions that extend beyond the pons. Herein, we report an unusual presentation of CPM that occurred secondary to a hyperglycaemic hyperosmolar state.
CASE REPORT
A 45-year-old woman presented to the emergency department with an unsteady gait, dizziness and right-sided weakness. Her medical history was significant for diabetes mellitus (with diabetic nephropathy), hypertension, hyperlipidaemia, total abdominal hysterectomy (for uterine fibroids) and pulmonary embolism. Her compliance to medications was suboptimal.
General physical examination was unremarkable. Neurological examination revealed that the patient was alert; cranial nerves were normal and power was grade 5 in all her limbs. She also had mild right-sided pronator drift and an ataxic gait. An initial diagnosis of posterior circulation stroke was made in the emergency department, and the patient was given a dose of aspirin. Laboratory investigations showed that the patient had significant hyperglycaemia (27.3 mmol/L), without ketonaemia or acidosis. Computed tomography (CT) of the brain, performed approximately one hour after presentation, showed bilateral thalamic, globus pallidus and dentate calcifications; the brainstem appeared normal. Intravenous insulin and normal saline infusion was commenced, and the patient was immediately transferred to the neurology service.
Detailed review by the neurologist eight hours after the initial presentation revealed a history of progressively increasing ataxia over two weeks; the ataxia was associated with dysarthria, clumsiness and involuntary crying. Neurological examination was significant for emotional lability, dysarthria, and gait ataxia with bilateral dysmetria. Motor examination showed normal tone, power grade 5 in all limbs, absent knee and ankle jerks, and flexor plantar response. Glove and stocking pattern loss to vibration was noted on sensory examination. Laboratory investigations are summarised in
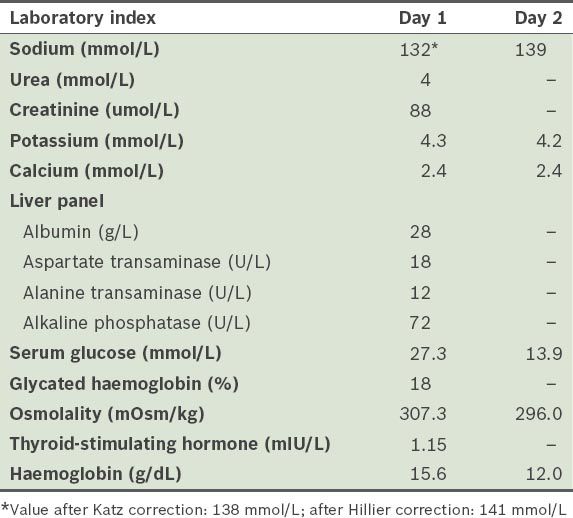
Table I
Laboratory indices of the patient.
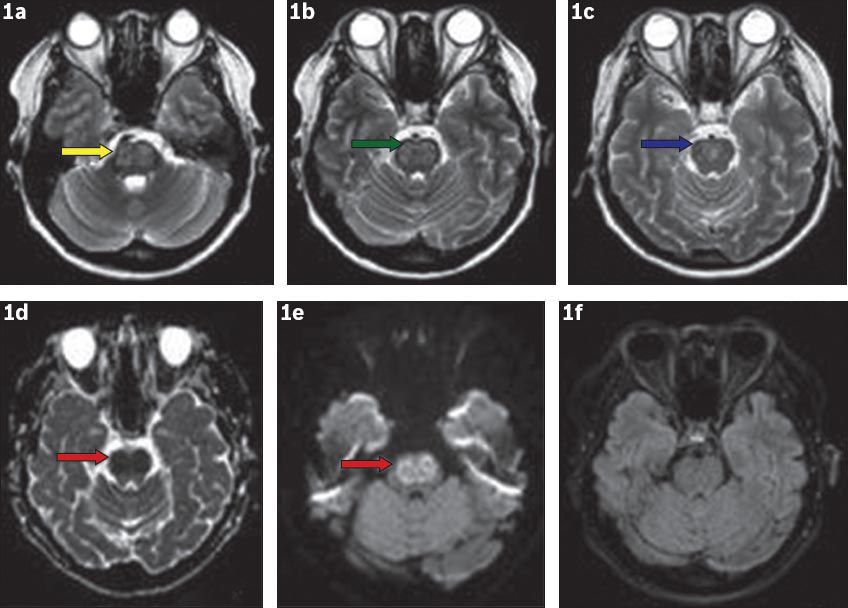
Magnetic resonance (MR) imaging was performed 14 hours after the initial CT. No progression or change in symptoms was reported by the patient during this interval, and clinical examination did not reveal any new deficits. MR imaging showed symmetrical, central T2-weighted (T2-W) hyperintensities in the pons and upper midbrain, and restricted diffusion on diffusion-weighted imaging sequences, with matched defect on the apparent diffusion coefficient map; the fluid attenuated inversion recovery images were normal (
Fig. 1
MR images of the brain show (a–c) symmetrical T2-W hyperintensities involving the pons (yellow/green arrow) and extending up to the lower midbrain (blue arrow). (d) Apparent diffusion coefficient image shows a hypointense lesion, which appears bright on (e) the diffusion-weighted image (red arrows). (f) Fluid attenuated inversion recovery image shows no abnormalities.
A diagnosis of CPM secondary to hyperglycaemia, with large-fibre peripheral diabetic neuropathy, was made. Diabetes mellitus management was optimised and physical rehabilitation was initiated. The patient was also prescribed escitalopram for the pseudobulbar affect. Gradual improvement in her gait was noted over the ensuing week. Brain imaging was not repeated in view of the patient’s clinical improvement and the absence of new neurological deficits.
DISCUSSION
In 1959, Adams et al first described CPM as a disease that affects alcoholics and the malnourished.(1) The classical presentation of the condition is sequential – initial encephalopathy or seizures, followed by rapid recovery in relation to electrolyte/osmolality correction, and subsequent clinical deterioration. Clinical manifestations include dysarthria, dysphagia, quadriparesis and alteration in the sensorium. Pupillary and oculomotor signs may also be noted. Extrapontine extension may occur, resulting in behavioural abnormalities and movement disorders.(1,2) In view of the expanded aetiological list and extrapontine manifestations, some authors prefer the term ‘osmotic demyelination’ to CPM.
Conditions associated with CPM include alcoholism, malnutrition, prolonged diuretic use, burns (in the context of hypernatraemia) and post-liver transplant.(3-6) CPM has also been described in patients with disturbances in serum osmolality in association with normal serum sodium.(5,7) In terms of hyperglycaemia, previous reports of CPM include cases with concomitant electrolyte disturbances or acidosis, or those occurring in relation to correction of the hyperglycaemic state.(7-10)
CPM is associated with myelin destruction in the centre of the basis pontis.(1) Extrapontine lesions may occur in the basal ganglia, thalamus and/or midbrain. Pathophysiologically, CPM is caused by a sudden shrinkage of the brain cells (particularly the oligodendrocytes), demyelination due to a rapid change in serum osmolality, and a shift in the idiogenic osmolytes.(1) The axons are largely preserved, except in advanced cases. Theories that have been postulated to explain these pathological changes include a disruption of the blood-brain barrier, resulting in localised demyelination and oligodendrocyte apoptosis.(11)
MR imaging changes that are associated with CPM include hyperintense symmetrical lesions on T2-W MR images, reflecting demyelination and oedema.(12) The transverse pontocerebellar fibres are most frequently involved, followed by the long rostrocaudal tracts; sparing of the tegmentum and corticospinal tract is classically seen. Diffusion-weighted imaging may detect lesions undetectable on T2-W MR images. Radiological mimics of CPM include demyelinating conditions (e.g. acute disseminated encephalomyelitis and multiple sclerosis), infarct and pontine neoplasms. Clinically, these conditions are differentiated by the time course of evolution, associated metabolic derangement, and specific radiological features such as enhancement and mass effect, in cases of neoplastic lesions.(12,13)
While the predilection for pontine involvement is the defining feature of CPM, the reason for this localisation remains unknown. The fact that the pons contains maximal admixture of grey and white matter elements has been hypothesised as a possible cause.(2) There is no specific treatment for CPM and management is largely supportive. Clinical features and/or the extent of radiological changes cannot reliably determine the prognosis. The outcome varies from recovery to a near-normal level of function to death.
The present case is unique in some aspects. First, a subacute presentation of CPM with predominant ataxia (reflecting involvement of the pontocerebellar fibres) and pseudobulbar affect (reflecting involvement of the corticobulbar tracts) is rare; our patient remained alert, with almost full preservation of her corticospinal function. Second, CPM was noted in context to hyperglycaemic hyperosmolality alone. Most previous reports of CPM occurring in the setting of hyperglycaemia describe concomitant ketoacidosis, abnormality in serum sodium, or occurrence after treatment of the hyperglycaemic hyperosmolar state. Burns et al reported the case of a patient who developed gait ataxia two days after receiving treatment for hyperosmolar hyperglycaemia,(8) while Guerrero et al reported the case of a young patient who developed hemiparesis and encephalopathy after being treated for hyperglycaemia.(10) O’Malley et al described the case of a female patient whose initial presentation was drowsiness with a plasma glucose level of 106 mmol/L; during treatment for hyperglycaemia, the patient experienced a rapid drop in plasma glucose (60 mmol/L drop within six hours, with flaccid quadraparesis and pseudobulbar palsy noted a few days after the treatment.(9) In the present case, although treatment for hyperglycaemia was initiated in our patient soon after her presentation, it is noteworthy that the detailed history obtained on review confirmed that there was slow evolution of her symptoms over the preceding two weeks. Furthermore, our patient did not report any change or new symptom between the time of her initial presentation to emergency service and MR imaging. Thus, we are inclined to believe that the observed radiological abnormalities, which conformed to the clinical presentation, were a result of the patient’s hyperglycaemic hyperosmolar state and not secondary to the treatment of the hyperglycaemic hyperosmolar state.
In summary, the present case report describes a rare presentation and aetiology of CPM. In addition to acute osmolality shifts, subacute changes may also underlie this condition. Hyperglycaemia should be included as an aetiology underlying CPM.