

Abstract
A 21-year-old woman presented with acute onset of upper abdominal pain. A diagnosis of Peutz-Jeghers syndrome (PJS) was made based on the clinical picture of perioral pigmentation with imaging findings of transient jejunojejunal intussusceptions and small bowel polyps, and confirmed by characteristic histopathological appearances of Peutz-Jeghers polyps. PJS is a rare hereditary condition characterised by unique hamartomatous polyps, perioral mucocutaneous pigmentations, and increased susceptibility to gastrointestinal and extraintestinal neoplasms. Patients usually present with recurrent abdominal pain due to intussusception caused by polyps. Other modes of presentations include rectal bleeding and melaena. We describe the imaging findings of PJS and provide a brief review of bowel polyposis syndromes. The latter are relatively rare disorders characterised by multiple polyps in the large or small intestine, with associated risk of malignancies and other extraintestinal manifestations. Awareness of the manifestations and early diagnosis of these syndromes is crucial to prevent further complications.
CASE PRESENTATION
A 21-year-old woman presented to the accident and emergency department with an acute onset of upper abdominal pain. The patient had similar and recurrent episodes of upper abdominal pain for the past six months. One month before this presentation, she had an episode of melaena with abdominal pain, which was clinically diagnosed as gastritis/bleeding ulcer and treated with anti-ulcer medications. On clinical examination, the patient was noted to have multiple lip pigmentations (
Fig. 1
Clinical photograph of the patient’s mouth.
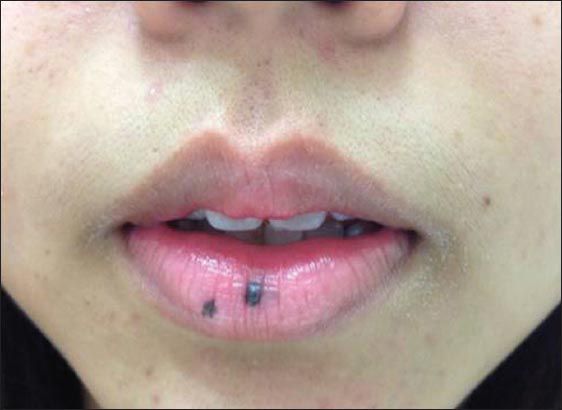
Fig. 2
Barium meal and follow-through image.
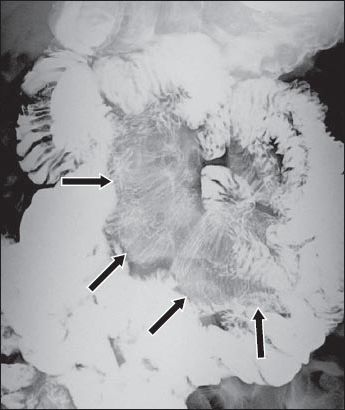
Fig. 3
(a) Axial and (b) coronal CT images of the abdomen.
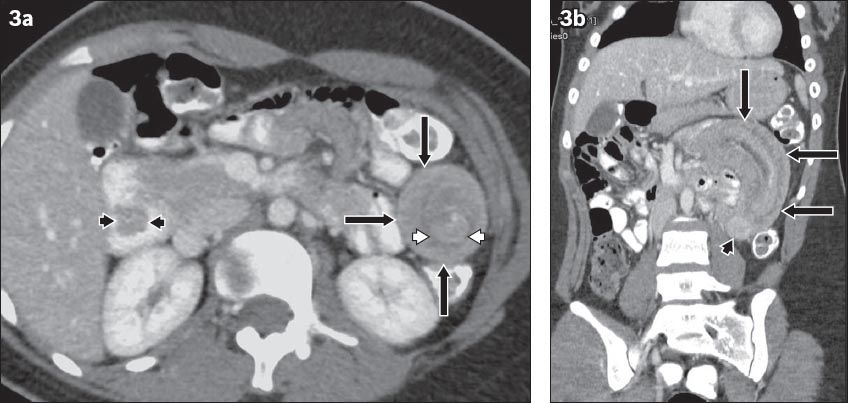
IMAGE INTERPRETATION
Barium meal and follow-through series (
DIAGNOSIS
Jejunal intussusception due to Peutz-Jeghers syndrome.
CLINICAL COURSE
Magnetic resonance enteroclysis (MRE) was performed on the patient one week after CT. MRE showed multiple jejunal and duodenal polyps (
Fig. 4
Coronal MRE images show (a & b) multiple jejunal polyps (arrows), and (c) a lobulated duodenal polyp (arrows).
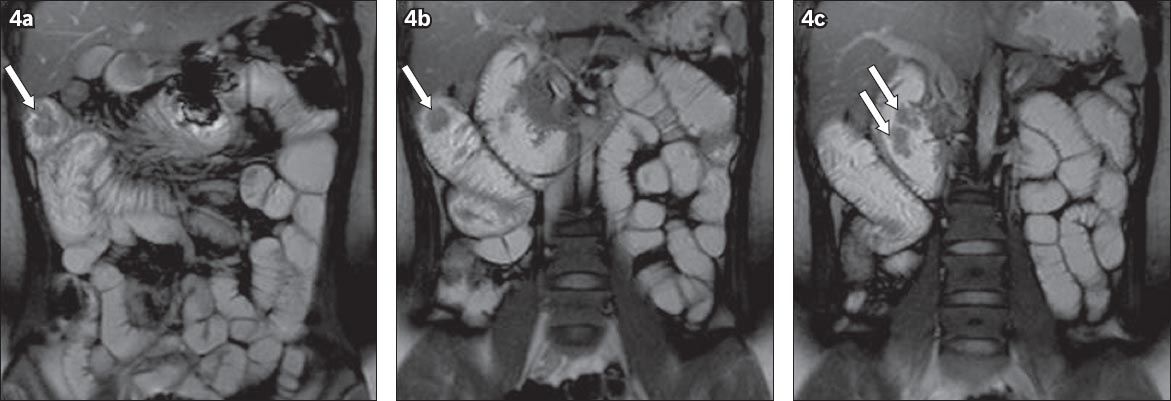
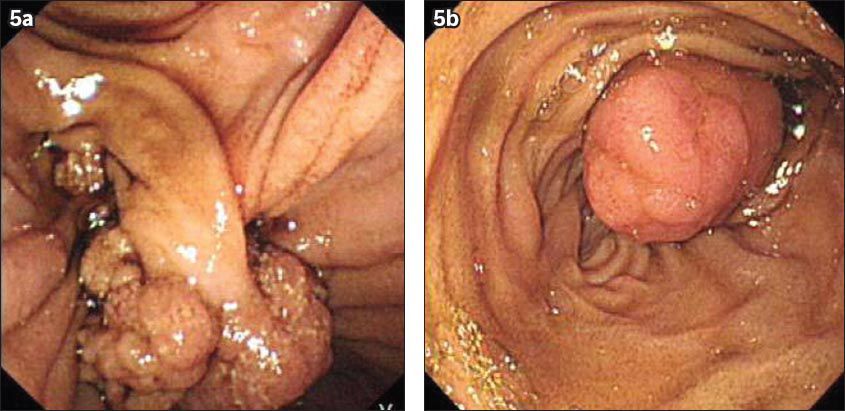
Fig. 5
Upper gastrointestinal endoscopic images show (a) a polyp in the second part of the duodenum, attached to the duodenal wall by a prolapsed short stalk, and (b) a polyp in the jejunum.
DISCUSSION
Peutz-Jeghers syndrome, first described by Peutz in 1921 and by Jegher in 1944, is characterised by hamartomatous polyps of the gastrointestinal tract (GIT) and mucocutaneous perioral pigmentation. It is an autosomal dominant inherited syndrome with incomplete penetrance, and its reported incidence is 1 in 150,000 births. Mutation of the tumour suppressor gene serine/threonine kinase 11, located on chromosome 19p13.3, is associated with Peutz-Jeghers syndrome. Spontaneous mutations give rise to some cases.(1-4) Lip and buccal mucosae are common locations for mucocutaneous pigmentations. The eyelid, dorsal aspect of the fingers and sole of the foot are less common locations. They are usually not seen at birth and appear by the first or second year of life. Skin and lip lesions disappear by adulthood, whereas buccal lesions remain.(5)
Peutz-Jeghers polyps can occur anywhere in the GIT except the oesophagus, and they are most commonly seen in the jejunum, followed by the ileum. The hamartomas in this syndrome are unique in that they have a characteristic smooth muscle core arising from the muscularis mucosae, which extends to the polyp. These polyps frequently present with recurrent abdominal pain due to intussusception. Intussusceptions often reduce spontaneously. They may cause intestinal obstruction if reduction does not occur. Ulceration of polyps may lead to bleeding or chronic anaemia. Other rare sites that develop polyps include the kidneys, ureter, nasal passages and bronchial tree.(4,6,7) Imaging usually shows multiple polyps and it is unusual to see only a solitary polyp. Large polyps have a lobulated appearance.(5) Polyps are often detected in barium studies, although they can also be seen in CT or ultrasonography (US). MRE can be used for the detection of polyps and has the advantage of a lack of ionising radiation. It has been shown that, for polyps measuring more than 10 mm in diameter, MRE has equal sensitivity to video capsule endoscopy (VCE).(8) Even though detection of small polyps is better with VCE, it cannot be used in the presence of obstruction.(9,10)
On radiography, intussusception may be seen as a soft tissue mass, commonly occupying the right upper quadrant. There may be a soft tissue mass containing concentric circular lucency (target sign) due to mesenteric fat in the intussusceptum. Another sign that may be identified is the meniscus sign, due to a crescent of air from the intussuscipiens outlining the intussusceptum at the apex of intussusception. Barium studies can demonstrate the meniscus sign (analogous to the radiograph) due to the projection of the intussusceptum into the barium-filled intussuscipiens at the apex. Barium may also reveal a ‘coiled-spring’ appearance of the bowel (i.e. coiled-spring sign) due to barium outlining the oedematous mucosal folds of the returning limb of the intussusceptum.(4,11) US typically reveals a ‘pseudokidney’ or ‘doughnut’ appearance, which consists of inner hyperechoic mesenteric fat that is dragged inside, between the entering and returning limbs of the intussusceptum and the outer hypoechoic rim, due to the oedematous wall of the intussuscipiens.(4,12) CT shows a ‘bowel-within-bowel’ appearance of the intussusceptions, seen as a sausage-shaped mass along the longitudinal axis of the bowel and a target-shaped mass perpendicular to the long axis of the bowel (
Patients with Peutz-Jegher syndrome have an increased risk of GIT malignancies, with an estimated risk of 2%–20%. The stomach, duodenum and colon are the most common locations. Even though hamartomas are commonly seen in the small intestine, no definite malignant transformation has been established. There is also an increased risk for extraintestinal malignancies, with a prevalence rate of 10%–30%. Common extraintestinal neoplasms include pancreatic, breast and reproductive organ (ovary, testis) tumours.(6) Small bowel polyposis syndromes include hamartomatous polyposis syndromes (where predominantly hamartomatous polyps are present), familial adenomatous polyposis (FAP) syndrome (where predominantly adenomatous polyps are present) and hereditary mixed polyposis syndrome. Conditions such as lymphoma (
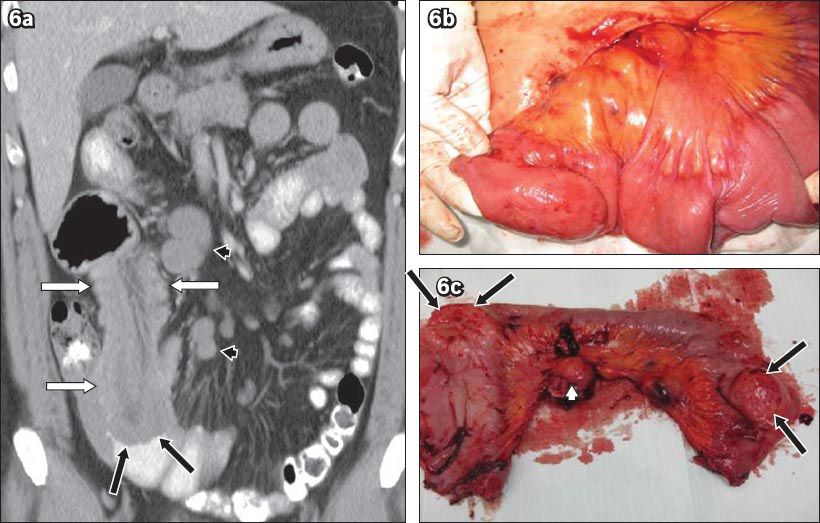
Fig. 6
Small bowel lymphoma with intussusception in a 50-year-old man who presented with colicky abdominal pain. (a) Coronal CT image shows small bowel intussusception (white arrows) and smooth-walled eccentric lesions in the small bowel (black arrows). Multiple enlarged mesenteric lymph nodes are present (arrowheads). The presence of homogeneously enhancing lymph nodes points to the imaging diagnosis of lymphoma. (b & c) Postoperative photographs of the resected specimen show two bulky small bowel lesions (arrows) and mesenteric lymph nodes (arrowhead). Histopathology of the lesions confirmed the diagnosis of lymphoma.
Hamartomatous polyposis syndromes (other than Peutz-Jeghers syndrome) include juvenile polyposis syndrome (JPS), Cowden syndrome, Bannayan-Riley-Ruvalcaba syndrome (BRRS), Proteus syndrome and Cronkhite-Canada syndrome.(1,6) An autosomal dominant disorder, JPS(1,6,14) is characterised by multiple hamartomatous polyps that can occur anywhere in the GIT except the oesophagus. The rectosigmoid colon is the most common location. Extraintestinal manifestations associated with this syndrome include cardiac defect, pulmonary arteriovenous malformation, polydactyly, clubbing, intestinal malrotation, Meckel’s diverticulum, macrocephaly, hypertelorism, cleft lip and palate, duplication of the renal pelvis and ureter, bifid uterus and vagina, undescended testes and supernumerary teeth. Patients with JPS have increased risk of colorectal, gastric and pancreatic carcinomas, and small intestinal adenocarcinoma.(1,14)
Cowden syndrome(6,15) is an autosomal dominant trait. Patients with this syndrome may have facial or oral papillomas, fibromas and skin tumours, in contrast to the perioral pigmentation seen in patients with Peutz-Jeghers syndrome. Other associated extraintestinal manifestations are abnormalities of the thyroid (goitre, adenoma), breast (fibrocystic disease, fibroadenoma) and central nervous system (CNS) (Lhermitte-Duclos disease). Cancers associated with this syndrome are follicular carcinoma of the thyroid, breast (ductal) carcinoma, uterine carcinoma, and transitional cell carcinoma of the renal pelvis and urinary bladder. BRRS(6,16) is an autosomal dominant syndrome characterised by multiple hamartomatous polyps of the small and large intestines, macrocephaly, pigmented lesions in the genital area, subcutaneous lipoma, visceral lipoma and haemangioma. Proteus syndrome(1,17) is a multisystem disorder characterised by hamartomas of multiple tissues, epidermal and connective tissue nevi, hemihypertrophy, hyperostosis and various tumours. Associated tumours include various testicular tumours, cystadenoma of the ovary, CNS tumours and monomorphic adenoma of the parotid gland. Cronkhite-Canada syndrome(6,18) differs from other polyposis syndromes in that it is not familial. In this syndrome, multiple small sessile polyps are noted throughout the GIT except the oesophagus. Patients may develop ectodermal abnormalities such as brownish macules on the palms and soles, and dystrophic nail changes. Cronkhite-Canada syndrome is associated with increased risk of colon and gastric cancers.(18)
FAP(19) and its variants are autosomal dominant disorders caused by mutation involving the adenomatous polyposis coli gene located on chromosome 5q21. Other variants of FAP include attenuated FAP (AFAP), Gardner’s syndrome and Turcot’s syndrome. In FAP (Figs.
Fig. 7
Familial adenomatous polyposis syndrome in a 32-year-old woman who presented with bouts of bloody diarrhoea. (a–c) Barium enema images show multiple smooth and rounded filling defects scattered throughout the colon (arrows). The patient underwent prophylactic total colectomy.
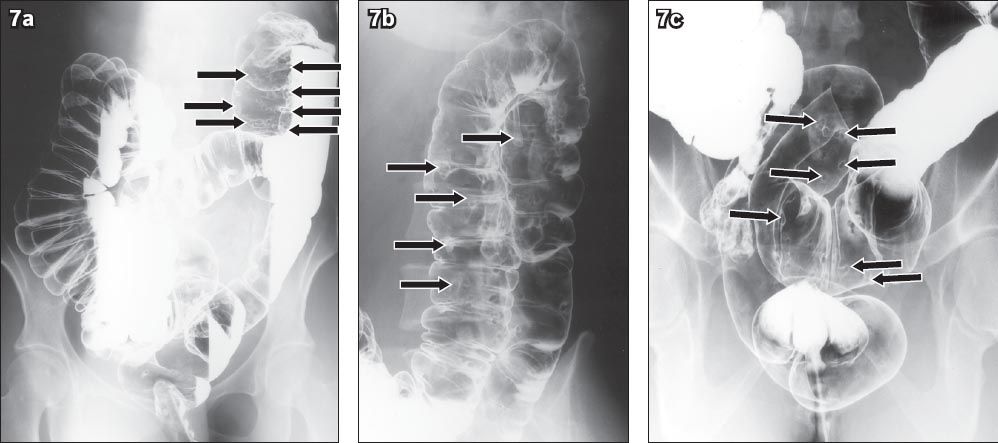
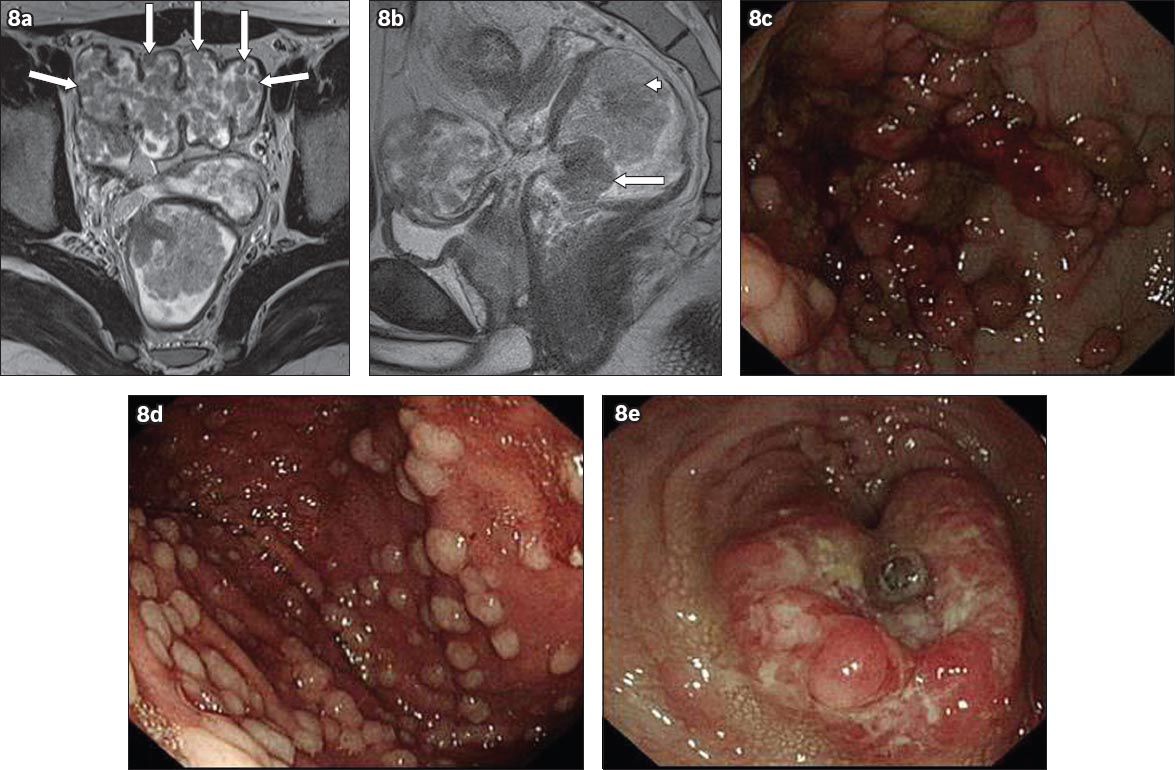
Fig. 8
Familial adenomatous polyposis syndrome in a 29-year-old man who was found to have rectal malignancy on routine follow-up. (a) Axial T2-W MR image shows multiple sigmoid polyps (arrows). (b) Sagittal T2-W MR image shows two large polypoidal lesions in the rectum (arrow) and rectosigmoid colon (arrowhead). Endoscopic images show multiple polyps in the (c) stomach; (d) colon; and (e) rectum. Biopsy of the rectal lesion confirmed an adenocarcinoma.
In summary, Peutz-Jeghers syndrome and other polyposis syndromes are associated with multiple GIT and extraintestinal malignancies. Knowledge of their clinical manifestations and early diagnosis of these syndromes is important so that early detection of future malignancies can be made through proper screening procedures, further complications may be avoided and appropriate management initiated.