Distribution of sasX, qacA/B and mupA genes and determination of genetic relatedness of methicillin-resistant Staphylococcus aureus among clinical isolates and nasal swab samples from the same patients in a hospital in Malaysia
Singapore Med J 2022; 63(6): 335-341 doi: 10.11622/smedj.2020166
Distribution of sasX, qacA/B and mupA genes and determination of genetic relatedness of methicillin-resistant Staphylococcus aureus among clinical isolates and nasal swab samples from the same patients in a hospital in Malaysia
Author Information >Copyright and License information >
1Department of Medical Microbiology and Parasitology, School of Medical Sciences, Kelantan, Malaysia 2Hospital USM, Health Campus, Universiti Sains Malaysia, Kelantan, Malaysia 3Acarology Unit, Infectious Disease Research Centre, Institute for Medical Research, Ministry of Health Malaysia, Selangor, Malaysia Correspondence: A/Prof Siti Asma’ Hassan, Consultant Clinical Microbiologist, Department of Medical Microbiology and Parasitology, School of Medical Sciences, Universiti Sains Malaysia, Health Campus, 16150 Kubang Kerian, Kelantan, Malaysia. sitiasmakb@usm.my
This study determined the distribution of sasX, qacA/B and mupA genes from methicillin-resistant Staphylococcus aureus (MRSA) isolated from clinical samples and nasal swab samples of the same patients and analysed their genetic relatedness.
METHODS
Polymerase chain reaction was used to detect the presence of sasX, qacA/B and mupA genes from 47 paired MRSA isolates. A paired isolate was defined as one nasal swab (colonising) isolate and clinical isolate that caused infection in the same patient. 22 selected paired isolates were subjected to multilocus sequence typing (MLST). The genetic relatedness among the isolates and association between the putative genes with epidemic sequence types (STs) were investigated.
RESULTS
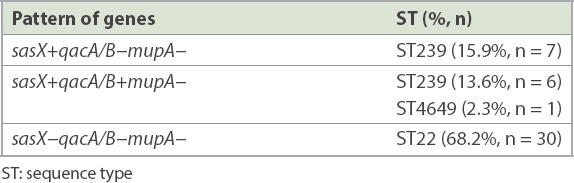
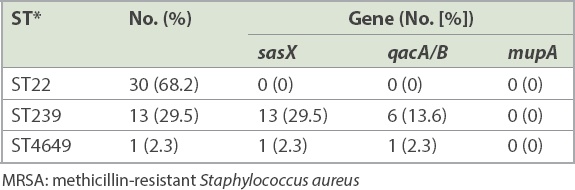
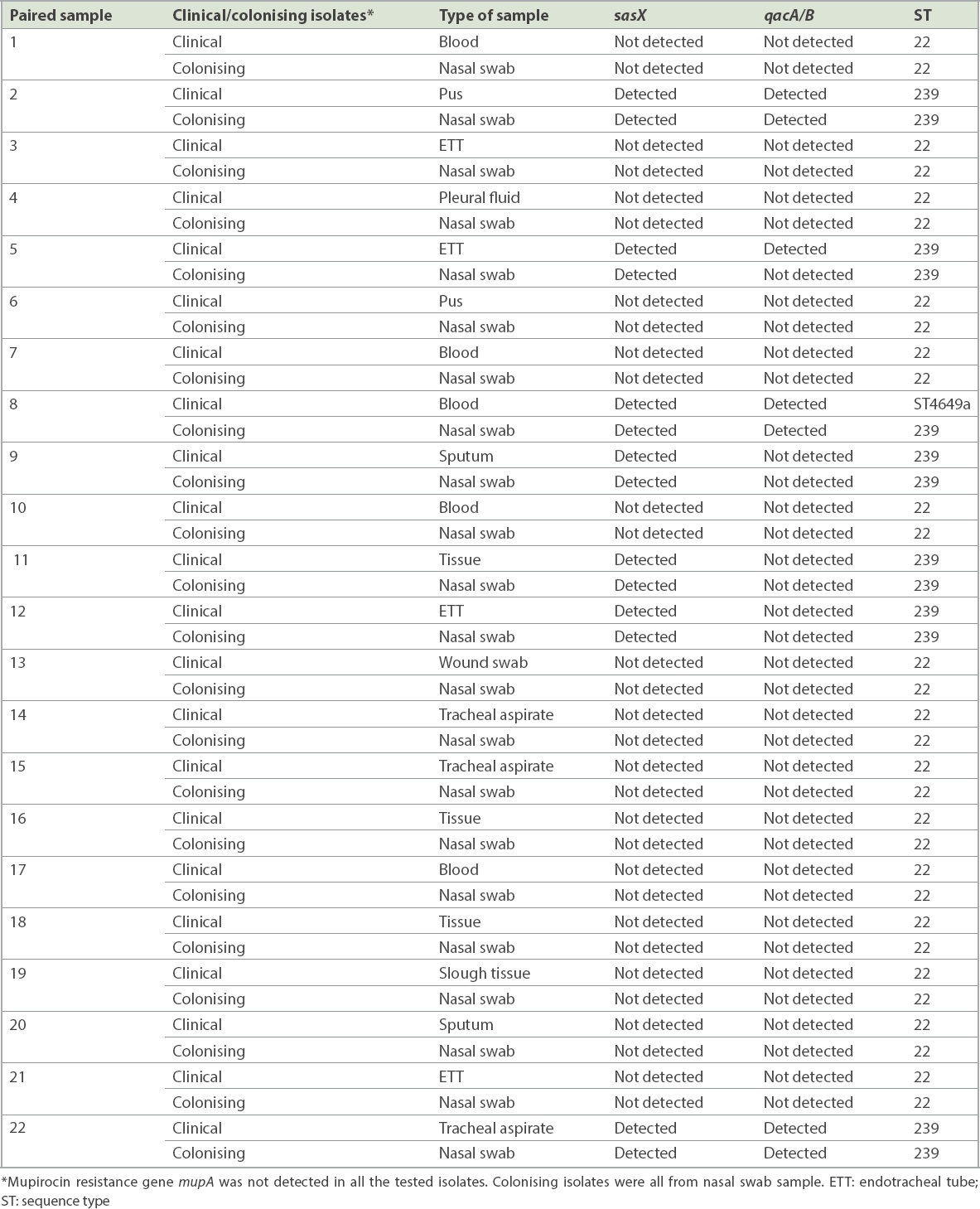
7 (14.9%, n = 14) paired isolates were positive for the sasX gene. qacA/B genes were positive in 7.4% (n = 7) of the isolates, from three paired isolates and one clinical isolate whose paired colonising isolate was negative. The paired sample of three patients were positive for both genes. The mupA gene was not detected in all the isolates. MLST revealed two epidemic STs, ST22 and ST239, and a novel ST4649. sasX and qacA/B genes were found in ST239 in 29.5% (n = 13) and 13.6% (n = 6) of cases, respectively. Gene co-existence occurred in 13.6% (n = 6) of MRSA ST239 and 2.3% (n = 1) of MRSA ST4649.
CONCLUSION
sasX and qacA/B genes were present in the MRSA isolates, while the mupA gene was undetected. ST22 and ST239 were the major MRSA clones. The circulating MRSA genotypes conferred different virulence and resistance determinants in our healthcare settings.
Keywords: clones, MRSA, nucleotide gene, sequence type
INTRODUCTION
Methicillin-resistant Staphylococcus aureus (MRSA) is a pathogen of global concern, causing both healthcare-associated and community-acquired infections. Anterior nares have been discovered as the main reservoir for MRSA.(1) Colonising strains may serve as endogenous reservoirs for overt clinical infections or may spread to other patients and lead to infection.(2) The emerging virulence factor of the sasX gene in MRSA strains may encourage MRSA colonisation in the anterior nares. sasX is among the newly described surface-anchored S. aureus-binding proteins that interface with human matrix molecules, MSCRAMMs (microbial surface components recognising adhesive matrix molecules).(3) Besides nasal colonisation, sasX contributes to biofilm formation and immune evasion mechanism. sasX-positive MRSA was reported to be a potential cause of serious diseases such as lung disease and abscess formation.(4) Chlorhexidine gluconate is one of the chemical agents used for decolonisation of MRSA.(5) The presence of the qacA/B gene is related to elevated minimum bactericidal concentrations for chlorhexidine and failures in MRSA decolonisation protocols.(6) Another drug that has been widely used for MRSA decolonisation is mupirocin ointment for local nasal application. The presence of the mupA gene is related to high mupirocin resistance and has been linked to the failure of mupirocin therapy for patients infected with MRSA.
The presence of both virulence and resistance genes in MRSA-specific clones has not been extensively evaluated. Hence, multilocus sequence typing (MLST) has been performed to study genetic relatedness in MRSA isolates from clinical specimens. One of the sequence types (STs) that was reported to have disseminated to other European countries and developed itself as the dominant clone is MRSA ST22. ST22 has replaced the Brazilian and Iberian clones, which were the predominant clones in Europe.(5)
MRSA ST239 is another strain that can produce exotoxins, causing a broad range of life-threatening infections.(7) MRSA ST239 is commonly associated with nosocomial infections and was also reported to be circulating in different countries with distinctive clone identities such as Hungarian, Brazilian, Portuguese and Viennese.(8) The detection of sasX, qacA/B and mupA genes, as well as the discovery of the circulating MRSA strains carrying specific genes, is significant for treatment plans in the healthcare setting and could intensify currently practised decontamination protocols.
METHODS
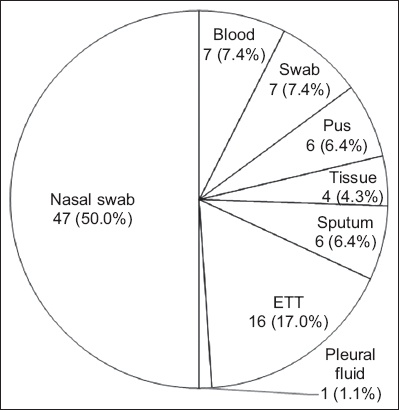
A total of 94 MRSA isolates (i.e. paired isolates of nasal swabs and clinical specimens of 47 patients) retrieved from archived isolates from the Microbiology Department, Hospital University Sains Malaysia, in Kelantan, Malaysia, from January 2016 to December 2017 were included in the study. A non-purposive sampling method was used. All confirmed, colonising and clinical MRSA isolates from the same patients within the study period were included. The isolates were from various specimens (Fig. 1).
Fig. 1
Chart shows the distribution of methicillin-resistant Staphylococcus aureus from 47 paired isolates. ETT: endotracheal tube
The isolates were reconfirmed using phenotypic methods such as Gram staining, lysis formation on blood agar and a few biochemical tests including catalase, deoxyribonuclease, tube coagulase and fermentation of mannitol. MRSA isolates were phenotypically confirmed using the cefoxitin susceptibility test according to the standard protocol established by USM Microbiology laboratory diagnostics, while inhibition zone diameter interpretation was based on Clinical and Laboratory Standards Institute (CLSI) 2017 criteria.(9) The amount of cefoxitin used was 30 mcg, with an inhibition zone of ≥ 22 mm. A minimum inhibitory concentration value of ≤ 4 mcg/mL was read as sensitive.
Genomic DNA extraction from clinical isolates was performed using the QIAamp DNeasy Blood and Tissue Kit (QIAGEN, Hilden, North Rhine-Westphalia, Germany), according to the manufacturer’s instructions, with slight modification. MRSA isolates were molecularly confirmed by polymerase chain reaction (PCR) amplification of femA and mecA genes.(10) ATCC 25923 and ATCC 33591 S. aureus strains were used as controls for femA and mecA gene detection, respectively.
Virulence and resistance genes were detected using published protocols.(11-13) Positive controls for sasX (MK509012) and qacA/B (MK542001) genes were obtained from local MRSA isolates. These isolates were characterised and confirmed by sequence analysis. ATCC BAA 1708 was used as the positive control for mupA gene detection according to M100, CLSI 2017 (28th edition).(9)
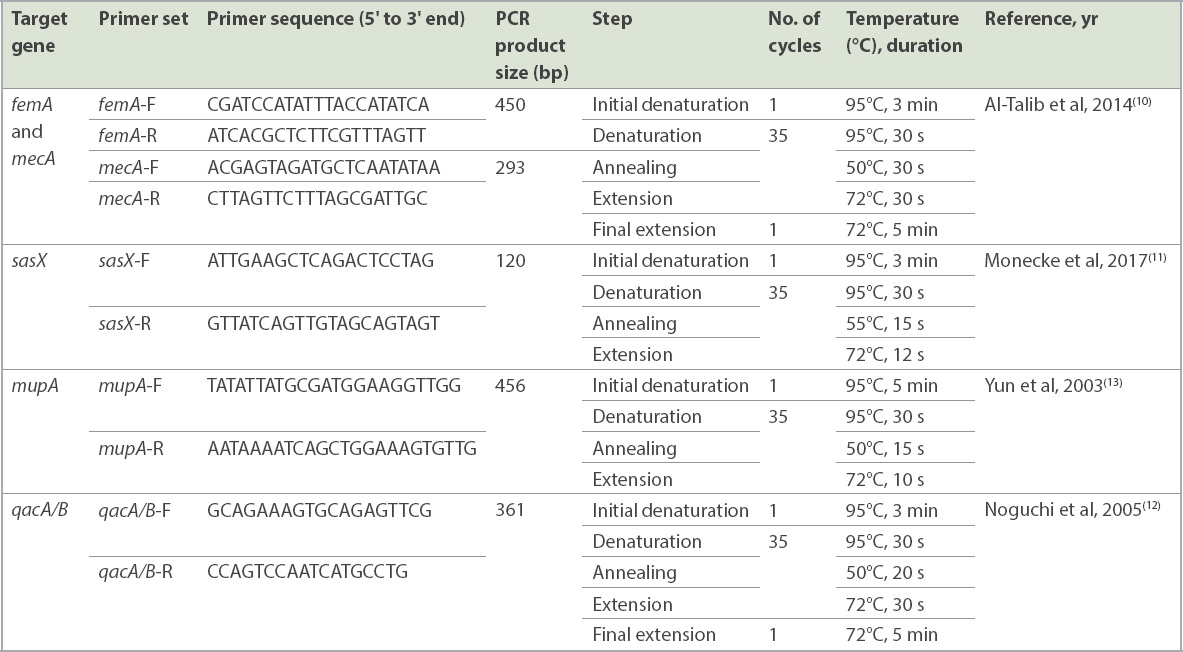
DNA amplification was performed in 25 mL of PCR mixture (Thermo Fisher Scientific, Vilnius, Vilniaus, Lithuania) consisting of 20 mM each of forward and reverse primers, 1 U/mL Taq Polymerase, 160 mM dNTP mix, PCR water, 10 × Taq buffer with (NH4)2-MgCl2, 25 mM MgCl2 and 2 mL of DNA template (20 ng/mL). The oligonucleotides primers used in this study have been previously described and were synthesised commercially (Integrated DNA Technologies, Singapore). The PCR reaction was performed using the Mastercycler nexus gradient thermal cycler (Eppendorf, Hamburg, Germany). PCR amplification for the respective targets was conducted using published primers (Table I). The gel was stained with Florosafe (Apical Scientific Sdn Bhd, Selangor, Malaysia) and visualised using a gel imaging system (AlphaImager; Alpha Innotec, Kasendorf, Germany).
Table I
List of primer sequences and polymerase chain reaction (PCR) thermal cycle conditions for each target gene.
A total of 22 paired MRSA isolates were selected for the MLST study based on sample types and clinical presentations. Seven S. aureus housekeeping genes (arcC, aroE, glpF, gmk, pta, tpi and yqiL) were amplified using the Mastercycler nexus gradient thermal cycler (Eppendorf). PCR reaction was performed with an initial denaturation at 95°C for five minutes, followed by 30 cycles of denaturation at 95°C for one minute, annealing at 55°C for one minute and extension at 72°C for one minute; and a final extension at 72°C for five minutes. All primer sequences and PCR protocols used were synthesised based on a previous study.(14) The protocol and PCR conditions for the respective genes used are shown in Table I. The PCR products were sent for sequencing analysis (Apical Scientific Sdn Bhd, Selangor, Malaysia), performed bidirectionally. Each sequence contig was submitted to the Staphylococcus aureus webpage on PubMLST (https://pubmlst.org/organisms/staphylococcus-aureus) for allelic profiles and ST characterisation. The phylogenetic tree was constructed using MEGA 7 software to demonstrate the distribution of STs and correlation of MRSA isolated from nasal swabs and clinical sources.(15)
Data was analysed using descriptive analysis. All graphs related to the study were constructed and described in terms of percentage and frequency. Ethical clearance was obtained and approval was granted for implementation by the Human Research Ethics Committee (JEPeM) of Universiti Sains Malaysia (study protocol code USM/JEPeM/17010056).
RESULTS
A total of 47 paired MRSA isolates were analysed. The distribution of the isolates from various specimens is shown in Fig. 1. PCR showed that 7 (14.9%, n = 14) paired isolates were positive for the sasX gene. Meanwhile, qacA/B genes were positive in 7.4% of the isolates, consisting of three paired isolates and one clinical isolate for which the respective paired colonising isolate was negative. The paired samples of three patients were positive for both genes. None of the isolates were positive for the mupA gene. Among 22 selected paired isolates that were subjected to MLST, three distinctive STs, namely ST22 (68.2%, n = 30), ST239 (29.5%, n = 13) and a novel ST, ST4649 (2.3%, n = 1), were identified. The gene distributions are shown in Tables II and III. The mupA gene was absent in all STs. Co-existence of the sasX and qacA/B genes occurred exclusively in 13.6% (n = 6) of MRSA ST239 and 2.3% (n = 1) of MRSA ST4649. The association of virulence-related, chlorhexidine resistance and mupirocin resistance genes with different STs of MRSA is shown in Table IV.
Table II
Distinctive sequence types with gene distribution patterns of 44 paired isolates.
Table III
Gene distribution of MRSA isolates in different sequence types.
Table IV
Association of virulence-related sasX gene, chlorhexidine resistance qacA/B gene and mupirocin resistance mupA gene with different sequence types of selected 22 paired MRSA isolates.
The phylogenetic relationship analysis revealed genetic variability among MRSA isolates, which were grouped into two major clades (Clade I and II), as shown in Fig. 2. Two predominant clones identified in this study were ST22 and ST239, while ST4649 was found to be novel, with a unique ST. The phylogenetic tree indicated that the MRSA isolates discovered from both nasal and clinical samples were clonal.
Fig. 2
Phylogenetic tree shows the genetic relatedness of paired isolates (n = 47). Paired samples were arranged according to the patient sequence in Table IV.
DISCUSSION
Patients who have MRSA nasal colonisation are at high risk of developing subsequent infections.(2) The most frequent MRSA isolation in our study was from endotracheal tube (ETT) fluid secretions (17.0%, n = 16). The pathogen was able to colonise ETTs and thrive in biofilm formed on the ETTs of intubated patients. This colonisation could increase the risk of developing ventilator-associated pneumonia following intubation, especially in intensive care units, and increase the mortality rates of hospitalised patients.(16) Aside from ETT secretions, 7.4% of the isolated MRSA were from blood samples and swab samples (n = 7). In a 2018 study, Lin et al reported that six out of ten patients had paired colonising and clinical isolates (wound culture) from MRSA, with one positive methicillin-sensitive S. aureus isolated from a patient.(17) Previous studies on MRSA colonisation causing subsequent infections demonstrated that the most common paired clinical isolates were from wounds or abscesses (48 out of 85)(18) and blood (31 out of 217).(19) However, the data is not comparable owing to differences in study design.
Involvement of various MRSA virulence genes potentially contributes to its infection severity and endemicity. In our study, 14.9% (n = 14) of the MRSA isolates carried the sasX gene. A study conducted in a China hospital detected the sasX gene in 36.7% of cases.(20) However, the reported outcomes cannot be used to compare with the current study owing to differences in study approach and sample size (94 vs. 610).(20) In the present study, the sasX gene was detected in isolates from various sample types such as wound swab, ETT secretions, blood, sputum, tracheal aspirate and tissue. One sasX gene was detected in ST4649, while others (n = 13) were exclusively from ST239. The finding paralleled those from other reports.(21,22) Despite the common occurrence of the sasX gene in ST239, Nair et al reported in 2013 that the sasX gene was not detected in all MRSA isolates, including MRSA ST239, from a Mongolian hospital. However, the contrasting finding might be attributed to its low prevalence in their hospital.(23)
The present study found qacA/B genes in 7.4% (n = 7) of the MRSA isolates. In contrast, a recent study conducted in Gansu Provincial Hospital, China, showed that all 85 MRSA isolates were positive for the qacA/B gene.(24) Another previous study also reported a high frequency (83.3%, 50 out of 60) of qacA/B genes in MRSA isolates collected from Malaysia.(25) The association between qacA/B genes and ST was also observed in this study. Our findings showed that qacA/B genes were found mostly in ST239 (13.6%, n = 6), with 2.3% (n = 1) in ST4649. This finding is supported by Lu et al’s 2014 report of 4.7% qacA/B-positive MRSA in ST239.(26) However, Ho et al earlier reported a contradictory finding, in which a high frequency of qacA/B genes (88.9%) was detected in ST239.(27)
A previous study reported the increased incidence of mupirocin resistance in MRSA and the failure of decolonisation treatments.(26) Fortunately, the mupA gene was not detected in all MRSA isolates of this study. A similar finding was reported by Nejabat et al.(28) However, a previous study conducted in Malaysia found that 70% (11 out of 16) of ST239 isolates were positive for the mupA gene.(29)
Our study showed the advantages of MLST data analysis in the determination of genetic variability among MRSA isolates, genetic relationship between nasal swab and clinical samples from MRSA strains, association between virulence and resistance genes with different STs, and genetic relatedness among the MRSA strains from neighbouring countries.
Phylogenetic analysis reported genetic diversity among MRSA isolates, with two major clades consisting of three different STs. The most frequent MRSA clone circulating in this healthcare setting was ST22 (68.2%, n = 30), which is similar to the findings of Espadinha et al in 2013.(30) The first isolation of ST22 in Malaysia was reported in 2009 and the emergence of multidrug-resistant ST22-MRSA-IV in a tertiary hospital in Malaysia was reported a few years later.(29) Thus, this study is significant, as it revealed that ST22 is gaining prominence in a Malaysian hospital.
The second predominant ST found in this study was ST239 (29.5%, n = 13). The high prevalence of MRSA ST239 in the Asian region might be attributed to the dissemination of a few epidemic clones. ST239-MRSA-III was a major MRSA clone found in Asian hospitals (i.e. healthcare-acquired MRSA), particularly in China and some Southeast Asian countries.(31) A study conducted by Sit et al in 2017 showed the predominance of ST239-MRSA-III circulating in a tertiary teaching hospital in Malaysia.(32)
The two major STs (ST22 and ST239) circulating in this hospital from different localities were nosocomial strains. This indicates that the infection control measures in the hospital setting should be further enhanced. Furthermore, the same major genotypes were reported in an Australian hospital.(32) However, in Singaporean hospitals, ST22 remained the predominant genotype in circulation. In addition, a new nosocomial strain, ST45, was reported to be gradually replacing ST239.(32,33) Hence, this study concluded that surveillance is essential for the monitoring and control of infectious diseases.
A novel ST was discovered in this study and assigned as ST4649. It is a single locus variant of ST239 that is distinguished by one mutation in the arcC gene allele. On the phylogenetic tree, ST4649 was clustered in the same clade as ST239, denoting their genetically close relationship. It is noteworthy that genetic diversity can be attributed to horizontal virulence gene transfer; the occurrence of novel STs reflects the ongoing dynamic process of genetic recombination and clonal expansion of the bacterial population.(32)
In terms of genetic relatedness, MRSA isolates from nasal swabs and clinical samples of 22 patients were highly related and clonal. Hence, nasal colonisation of MRSA might subsequently lead to infection. A similar study has demonstrated that two major STs, ST239 and ST5, had a genetic correlation between nasal and clinical isolates.(34) Studies by Tilahun et al and Kao et al also revealed that the same MRSA genotypes originated from both colonisation and infection sites.(35,36)
Based on the association of virulence and resistance genes in different ST and their distribution patterns, this study revealed the co-existence of sasX and qacA/B genes in MRSA isolates. Gene co-existence occurred exclusively in 13.6% (n = 6) of ST239 and merely 2.3% (n = 1) of ST4649, which was comparable to previous studies.(20,37) To the best of our knowledge, this is the first report of the co-existence of sasX- and qacA/B-positive ST239 in a Malaysian tertiary hospital.
This study was not without limitations. Correlation validation was not performed between the laboratory results and the clinical outcomes. Hence, the correlation between clinical outcomes and the study outcome provides a good prospect for future research. As molecular typing using MLST was performed only on selective MRSA strains, molecular characterisation of entire MRSA strains may describe the high genetic variability of the MRSA being studied and the higher frequency of genetic relatedness between nasal and clinical isolates.
In conclusion, the sasX gene and qacA/B gene were present in MRSA isolates in our healthcare setting, while the mupA gene was not detected. ST22 and ST239 were the major MRSA clones that predominated our healthcare setting, and a new ST4649 was discovered. We have demonstrated that the circulating MRSA genotypes conferred different virulence and resistance determinants.
ACKNOWLEDGEMENTS
This study was funded by the Usains Research Grant 2016 (401.PPSP.3410015) as well as the Tabung Insentif Pembangunan Pengajian Siswazah PPSP 2017 (308.AIPPSP.415403). We would also like to thank Mrs Silamai A/P Ea Chum and Dr Wan Nor Arifin for their help in research and statistical analysis and Mr Ahmad Adebayo Irekeola for English editing.
References Abad CL, Pulia MS, Safdar N.Does the nose know?An update on MRSA decolonization strategies.Curr Infect Dis Rep. 2013;15:455-64. Albrecht VS, Limbago BM, Moran GJ, et al. Staphylococcus aureus colonization and strain type at various body sites among patients with a closed abscess and uninfected controls at US emergency departments.JClin Microbiol. 2015;53:3478-84. Li M, Du X, Villaruz AE, et al. MRSA epidemic linked to a quickly spreading colonization and virulence determinant.Nat Med. 2012;18:816-9. Dhiman M, Bhatt N, Chau-Han V, Farooq U, Khan A.Emergence of methicillin resistantStaphylococcus aureus(MRSA) isolates from north India harboring a novel sasX gene:further analyzing its role in biofilm formation.Ann Med Biomed Sci. 2017;3:46-50. Aires de Sousa MA, Sanches IS, Ferro ML, et al. Intercontinental spread of a multidrug-resistant methicillin-resistant staphylococcus aureus clone.JClin Microbiol. 1998;36:2590-6. Warren DK, Prager M, Munigala S, et al. Prevalence of qacA/B genes and mupirocin resistance among methicillin-resistant Staphylococcus aureus(MRSA) isolates in the setting of chlorhexidine bathing without mupirocin.Infect Control Hosp Epidemiol. 2016;37:590-7. Abimanyu N, Murugesan S, Krishnan P.Emergence of methicillin-resistant Staphylococcus aureusST239 with high-level mupirocin and inducible clindamycin resistance in a tertiary care center in Chennai, South India.J Clin Microbiol. 2012;50:3412-3. Feil EJ, Nickerson EK, Chantratita N, et al. Rapid detection of the pandemic methicillin-resistantStaphylococcus aureus clone ST 239, a dominant strain in Asian hospitals.J Clin Microbiol. 2008;46:1520-2. Clinical and Laboratory Standards InstitutePerformance standards for antimicrobial susceptibility testing. 27th edition. CLSI supplement M100. 2017;Pennsylvannia Clinical and Laboratory Standards Institute. Al-Talib H, Yean CY, Al-Khateeb A, Hasan H, Ravichandran M.Rapid detection of methicillin-resistantStaphylococcus aureus by a newly developed dry reagent-based polymerase chain reaction assay.J Microbiol Immunol Infect. 2014;47:484-90. Monecke S, Müller E, Dorneanu OS, VremerÄ T, Ehricht R.Molecular typing of MRSA and of clinical Staphylococcus aureus isolates from IaÅi, Romania.PLoS One. 2014;9:e97833. Noguchi N, Suwa J, Narui K, et al. Susceptibilities to antiseptic agents and distribution of antiseptic-resistance genes qacA/B and smr of methicillin-resistant Staphylococcus aureus isolated in Asia dang, 1998 and 1999.JMed Microbiol. 2005;54:Pt 6557-65. Yun HJ, Lee SW, Yoon GM, et al. Prevalence and mechanisms of low- and high-level mupirocin resistance in staphylococci isolated from a Korean hospital.JAntimicrob Chemother. 2003;51:619-23. Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG.Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus.JClin Microbiol. 2000;38:1008-15. Kumar S, Stecher G, Li M, Knyaz C, Tamura K.MEGA X:Molecular Evolutionary Genetics Analysis across Computing Platforms.Mol Biol Evol. 2018;35:1547-49. Percival SL, Suleman L, Vuotto C, Donelli G.Healthcare-associated infections, medical devices and biofilms:risk, tolerance and control.J Med Microbiol. 2015;64:323-34. Lin SY, Lin NY, Huang YY, Hsieh CC, Huang YC.Methicillin-resistant Staphylococcus aureus nasal carriage and infection among patients with diabetic foot ulcer.J Microbiol Immunol Infect. 2018;pii:S1684-1182(18)30155-5. Eko KE, Forshey BM, Carrel M, et al. Molecular characterization of methicillin-resistant Staphylococcus aureus (MRSA) nasal colonization and infection isolates in a Veterans Affairs hospital.Antimicrob Resist Infect Control. 2015;4:10. Robicsek A, Suseno M, Beaumont JL, Thomson RB, JrPeterson LR.Prediction of methicillin-resistant Staphylococcus aureus involvement in disease sites by concomitant nasal sampling.JClin Microbiol. 2008;46:588-92. Song Y, Du X, Li T, Zhu Y, Li M.Phenotypic and molecular characterization of Staphylococcus aureusrecovered from different clinical specimens of inpatients at a teaching hospital in Shanghai between 2005 and 2010.J Med Microbiol. 2013;62:Pt 2274-82. Eshaghi M, Bibalan M, Pournajaf A, Gholami M, Talebi M.Detection of new virulence genes in mecA-positiveStaphylococcus aureus isolated from clinical samples:the first report from Iran.Infect Dis Clin Pract. 2017;25:310-3. Zarizal S, Yeo CC, Faizal GM, et al. Nasal colonisation, antimicrobial susceptibility and genotypic pattern of Staphylococcus aureusamong agricultural biotechnology students in Besut, Terengganu, east coast of Malaysia.Trop Med Int Health. 2018;23:905-13. Nair R, Hanson BM, Kondratowicz K, et al. Antimicrobial resistance and molecular epidemiology of Staphylococcus aureus from Ulaanbaatar, Mongolia.PeerJ. 2013;1:e176. Chen X, Wu Z, Zhou Y, et al. Molecular and virulence characteristics of methicillin-resistant Staphylococcus aureusin burn patients.Front Lab Med. 2017;1:43-7. Shamsudin MN, Alreshidi MA, Hamat RA, et al. High prevalence of qacA/B carriage among clinical isolates of meticillin-resistantStaphylococcus aureusin Malaysia.J Hosp Infect. 2012;81:206-8. Lu Z, Chen Y, Chen W, et al. Characteristics of qacA/B-positive Staphylococcus aureusisolated from patients and a hospital environment in China.J Antimicrob Chemother. 2014;70:653-7. Ho J, Branley J.Prevalence of antiseptic resistance genes qacA/B and specific sequence types of methicillin-resistantStaphylococcus aureus in the era of hand hygiene.JAntimicrob Chemother. 2012;67:1549-50. Nejabat M, Khashei R, Bazargani A, Ebrahim-Saraie HS, Motamedifar M.Evaluation of high-level of mupirocin resistance among clinical isolates of methicillin-resistantStaphylococcus aureusfrom Shiraz, Iran (2008-2009).Pharm Sci. 2015;21:225-8. Ghasemzadeh-Moghaddam H, van Belkum A, Hamat RA, van Wamel W, Neela V.Methicillin-susceptible and -resistant Staphylococcus aureus with high-level antiseptic and low-level mupirocin resistance in Malaysia.Microb Drug Resist. 2014;20:472-7. Espadinha D, Faria NA, Miragaia M, et al. Médicos Sentinela Network. Extensive dissemination of methicillin-resistant Staphylococcus aureus (MRSA) between the hospital and the community in a country with a high prevalence of nosocomial MRSA.PLoS One. 2013;8:e59960. Chen CJ, Huang YC.New epidemiology ofStaphylococcus aureusinfection in Asia.Clin Microbiol Infect. 2014;20:605-23. Sit PS, Teh CS, Idris N, et al. Prevalence of methicillin-resistant Staphylococcus aureus(MRSA) infection and the molecular characteristics of MRSA bacteraemia over a two-year period in a tertiary teaching hospital in Malaysia.BMC Infect Dis. 2017;17:274. Teo J, Tan TY, Hon PY, et al. Network for Antimicrobial Resistance Surveillance (Singapore). ST22 and ST239 MRSA duopoly in Singaporean hospitals:2006-2010.Epidemiol Infect. 2013;141:153-7. Jeremiah C, Kandiah JP, Spelman DW, et al. Differing epidemiology of two major healthcare-associated meticillin-resistant Staphylococcus aureusclones.JHosp Infect. 2016;92:183-90. Tilahun B, Faust AC, McCorstin P, Ortegon A.Nasal colonization and lower respiratory tract infections with methicillin-resistantStaphylococcus aureus.Am J Crit Care. 2015;24:8-12. Kao KC, Chen CB, Hu HC, et al. Risk factors of methicillin-resistant Staphylococcus aureus infection and correlation with nasal colonization based on molecular genotyping in medical intensive care units:a prospective observational study.Medicine (Baltimore). 2015;94:e1100. Kaczmarek M, Mullett MS, McDonald JE, Denman S.Multilocus sequence typing provides insights into the population structure and evolutionary potential of Brenneria goodwinii, associated with acute oak decline.PLoS One. 2017;12:e0178390.
REFERENCES
1. Abad CL, Pulia MS, Safdar N. Does the nose know? An update on MRSA decolonization strategies. Curr Infect Dis Rep 2013; 15:455-64. https://doi.org/10.1007/s11908-013-0364-y
PMid:24150839 PMCid:PMC4258870
2. Albrecht VS, Limbago BM, Moran GJ, et al. Staphylococcus aureus colonization and strain type at various body sites among patients with a closed abscess and uninfected controls at US emergency departments. J Clin Microbiol 2015; 53:3478-84. https://doi.org/10.1128/JCM.01371-15
PMid:26292314 PMCid:PMC4609677
3. Li M, Du X, Villaruz AE, et al. MRSA epidemic linked to a quickly spreading colonization and virulence determinant. Nat Med 2012; 18:816-9. https://doi.org/10.1038/nm.2692
PMid:22522561 PMCid:PMC3378817
4. Dhiman M, Bhatt N, Chau-Han V, Farooq U, Khan A. Emergence of methicillin resistant Staphylococcus aureus (MRSA) isolates from north India harboring a novel sasX gene: further analyzing its role in biofilm formation. Ann Med Biomed Sci 2017; 3:46-50.
5. Aires de Sousa MA, Sanches IS, Ferro ML, et al. Intercontinental spread of a multidrug-resistant methicillin-resistant staphylococcus aureus clone. J Clin Microbiol 1998; 36:2590-6. https://doi.org/10.1128/JCM.36.9.2590-2596.1998
PMid:9705398 PMCid:PMC105168
6. Warren DK, Prager M, Munigala S, et al. Prevalence of qacA/B genes and mupirocin resistance among methicillin-resistant Staphylococcus aureus (MRSA) isolates in the setting of chlorhexidine bathing without mupirocin. Infect Control Hosp Epidemiol 2016; 37:590-7. https://doi.org/10.1017/ice.2016.1
PMid:26828094 PMCid:PMC4833605
7. Abimanyu N, Murugesan S, Krishnan P. Emergence of methicillin-resistant Staphylococcus aureus ST239 with high-level mupirocin and inducible clindamycin resistance in a tertiary care center in Chennai, South India. J Clin Microbiol 2012; 50:3412-3. https://doi.org/10.1128/JCM.01663-12
PMid:22855516 PMCid:PMC3457429
8. Feil EJ, Nickerson EK, Chantratita N, et al. Rapid detection of the pandemic methicillin-resistant Staphylococcus aureus clone ST 239, a dominant strain in Asian hospitals. J Clin Microbiol 2008; 46:1520-2. https://doi.org/10.1128/JCM.02238-07
PMid:18234867 PMCid:PMC2292922
9. Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing. 27th edition. CLSI supplement M100. Pennsylvannia: Clinical and Laboratory Standards Institute, 2017.
10. Al-Talib H, Yean CY, Al-Khateeb A, Hasan H, Ravichandran M. Rapid detection of methicillin-resistant Staphylococcus aureus by a newly developed dry reagent-based polymerase chain reaction assay. J Microbiol Immunol Infect 2014; 47:484-90. https://doi.org/10.1016/j.jmii.2013.06.004
PMid:23927820
11. Monecke S, Müller E, Dorneanu OS, Vremeră T, Ehricht R. Molecular typing of MRSA and of clinical Staphylococcus aureus isolates from Iaşi, Romania. PLoS One 2014; 9:e97833.
12. Noguchi N, Suwa J, Narui K, et al. Susceptibilities to antiseptic agents and distribution of antiseptic-resistance genes qacA/B and smr of methicillin-resistant Staphylococcus aureus isolated in Asia during 1998 and 1999. J Med Microbiol 2005; 54(Pt 6):557-65.
13. Yun HJ, Lee SW, Yoon GM, et al. Prevalence and mechanisms of low- and high-level mupirocin resistance in staphylococci isolated from a Korean hospital. J Antimicrob Chemother 2003; 51:619-23. https://doi.org/10.1093/jac/dkg140
PMid:12615863
14. Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol 2000; 38:1008-15. https://doi.org/10.1128/JCM.38.3.1008-1015.2000
PMid:10698988 PMCid:PMC86325
15. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol 2018; 35:1547-49. https://doi.org/10.1093/molbev/msy096
PMid:29722887 PMCid:PMC5967553
16. Percival SL, Suleman L, Vuotto C, Donelli G. Healthcare-associated infections, medical devices and biofilms: risk, tolerance and control. J Med Microbiol 2015; 64:323-34. https://doi.org/10.1099/jmm.0.000032
PMid:25670813
17. Lin SY, Lin NY, Huang YY, Hsieh CC, Huang YC. Methicillin-resistant Staphylococcus aureus nasal carriage and infection among patients with diabetic foot ulcer. J Microbiol Immunol Infect 2018; pii: S1684-1182(18)30155-5.
18. Eko KE, Forshey BM, Carrel M, et al. Molecular characterization of methicillin-resistant Staphylococcus aureus (MRSA) nasal colonization and infection isolates in a Veterans Affairs hospital. Antimicrob Resist Infect Control 2015; 4:10. https://doi.org/10.1186/s13756-015-0048-5
PMid:25838886 PMCid:PMC4383227
19. Robicsek A, Suseno M, Beaumont JL, Thomson RB Jr, Peterson LR. Prediction of methicillin-resistant Staphylococcus aureus involvement in disease sites by concomitant nasal sampling. J Clin Microbiol 2008; 46:588-92. https://doi.org/10.1128/JCM.01746-07
PMid:18057132 PMCid:PMC2238094
20. Song Y, Du X, Li T, Zhu Y, Li M. Phenotypic and molecular characterization of Staphylococcus aureus recovered from different clinical specimens of inpatients at a teaching hospital in Shanghai between 2005 and 2010. J Med Microbiol 2013; 62(Pt 2):274-82.
21. Eshaghi M, Bibalan M, Pournajaf A, Gholami M, Talebi M. Detection of new virulence genes in mecA-positive Staphylococcus aureus isolated from clinical samples: the first report from Iran. Infect Dis Clin Pract 2017; 25:310-3. https://doi.org/10.1097/IPC.0000000000000506
22. Zarizal S, Yeo CC, Faizal GM, et al. Nasal colonisation, antimicrobial susceptibility and genotypic pattern of Staphylococcus aureus among agricultural biotechnology students in Besut, Terengganu, east coast of Malaysia. Trop Med Int Health 2018; 23:905-13. https://doi.org/10.1111/tmi.13090
PMid:29873865
23. Nair R, Hanson BM, Kondratowicz K, et al. Antimicrobial resistance and molecular epidemiology of Staphylococcus aureus from Ulaanbaatar, Mongolia. PeerJ 2013; 1:e176.
24. Chen X, Wu Z, Zhou Y, et al. Molecular and virulence characteristics of methicillin-resistant Staphylococcus aureus in burn patients. Front Lab Med 2017; 1:43-7. https://doi.org/10.1016/j.flm.2017.02.010
25. Shamsudin MN, Alreshidi MA, Hamat RA, et al. High prevalence of qacA/B carriage among clinical isolates of meticillin-resistant Staphylococcus aureus in Malaysia. J Hosp Infect 2012; 81:206-8. https://doi.org/10.1016/j.jhin.2012.04.015
PMid:22633074
26. Lu Z, Chen Y, Chen W, et al. Characteristics of qacA/B-positive Staphylococcus aureus isolated from patients and a hospital environment in China. J Antimicrob Chemother 2014; 70:653-7. https://doi.org/10.1093/jac/dku456
PMid:25429780
27. Ho J, Branley J. Prevalence of antiseptic resistance genes qacA/B and specific sequence types of methicillin-resistant Staphylococcus aureus in the era of hand hygiene. J Antimicrob Chemother 2012; 67:1549-50. https://doi.org/10.1093/jac/dks035
PMid:22334609
28. Nejabat M, Khashei R, Bazargani A, Ebrahim-Saraie HS, Motamedifar M. Evaluation of high-level of mupirocin resistance among clinical isolates of methicillin-resistant Staphylococcus aureus from Shiraz, Iran (2008-2009). Pharm Sci 2015; 21:225-8. https://doi.org/10.15171/PS.2015.41
29. Ghasemzadeh-Moghaddam H, van Belkum A, Hamat RA, van Wamel W, Neela V. Methicillin-susceptible and -resistant Staphylococcus aureus with high-level antiseptic and low-level mupirocin resistance in Malaysia. Microb Drug Resist 2014; 20:472-7. https://doi.org/10.1089/mdr.2013.0222
PMid:24841796
30. Espadinha D, Faria NA, Miragaia M, et al; Médicos Sentinela Network. Extensive dissemination of methicillin-resistant Staphylococcus aureus (MRSA) between the hospital and the community in a country with a high prevalence of nosocomial MRSA. PLoS One 2013; 8:e59960.
31. Chen CJ, Huang YC. New epidemiology of Staphylococcus aureus infection in Asia. Clin Microbiol Infect 2014; 20:605-23. https://doi.org/10.1111/1469-0691.12705
PMid:24888414
32. Sit PS, Teh CS, Idris N, et al. Prevalence of methicillin-resistant Staphylococcus aureus (MRSA) infection and the molecular characteristics of MRSA bacteraemia over a two-year period in a tertiary teaching hospital in Malaysia. BMC Infect Dis 2017; 17:274. https://doi.org/10.1186/s12879-017-2384-y
PMid:28407796 PMCid:PMC5390426
33. Teo J, Tan TY, Hon PY, et al; Network for Antimicrobial Resistance Surveillance (Singapore). ST22 and ST239 MRSA duopoly in Singaporean hospitals: 2006-2010. Epidemiol Infect 2013; 141:153-7. https://doi.org/10.1017/S0950268812000337
PMid:22394568 PMCid:PMC9152064
34. Jeremiah C, Kandiah JP, Spelman DW, et al. Differing epidemiology of two major healthcare-associated meticillin-resistant Staphylococcus aureus clones. J Hosp Infect 2016; 92:183-90. https://doi.org/10.1016/j.jhin.2015.10.023
PMid:26778134
35. Tilahun B, Faust AC, McCorstin P, Ortegon A. Nasal colonization and lower respiratory tract infections with methicillin-resistant Staphylococcus aureus. Am J Crit Care 2015; 24:8-12. https://doi.org/10.4037/ajcc2015102
PMid:25554549
36. Kao KC, Chen CB, Hu HC, et al. Risk factors of methicillin-resistant Staphylococcus aureus infection and correlation with nasal colonization based on molecular genotyping in medical intensive care units: a prospective observational study. Medicine (Baltimore) 2015; 94:e1100.
37. Kaczmarek M, Mullett MS, McDonald JE, Denman S. Multilocus sequence typing provides insights into the population structure and evolutionary potential of Brenneria goodwinii, associated with acute oak decline. PLoS One 2017; 12:e0178390.