

Abstract
INTRODUCTION
The objectives of this study were to examine the effects of ethnicity, gender and a proton pump inhibitor (PPI), omeprazole, on the human gut microbiome. PPIs are commonly used for the treatment of acid-related disorders. We hypothesised that PPI therapy might perturb microbial communities and alter the gut microbiome.
METHODS
Healthy subjects of Chinese (n = 12), Malay (n = 12) and Indian (n = 10) ancestry, aged 21–37 years, were enrolled. They provided a baseline stool sample (Day 1) and were then given a course of omeprazole at therapeutic dose (20 mg daily) for seven days. Stool samples were collected again on Day 7 and 14 (one week after stopping omeprazole). Microbial DNA was extracted from the stool samples, followed by polymerase chain reaction, library construction, 16S rRNA sequencing using Illumina MiSeq, and statistical and bioinformatics analyses.
RESULTS
The findings showed an increase in species richness (p = 0.018) after omeprazole consumption on Day 7, which reverted to baseline on Day 14. There were significant increases in the relative abundance of Streptococcus vestibularis (p = 0.0001) and Veillonella dispar (p = 0.0001) on Day 7, which diminished on Day 14. Faecalibacterium prausnitzii, Sutterella stercoricanis and Bacteroides denticanum were characteristic of Chinese, Malays and Indians, respectively. Lactobacillaceae and Bacteroides xylanisolvens were the signature taxa of male and female subjects, respectively.
CONCLUSION
The study demonstrated alterations in the gut microbiome following omeprazole treatment. This may explain the underlying pathology of increased risk of Clostridium difficile infections associated with omeprazole therapy.
INTRODUCTION
The human microbiota refers to the collection of microbes that exist in the human body.(1) Research on microbial interactions with humans has generally focused on single pathogenic organisms and is a hypothesis-driven approach guided by prior knowledge. This is further constrained by our limited knowledge of the roles played by these microbes in physiology and pathology. Few studies have been conducted to profile communities of non-pathogenic microbes in the body, as they were thought to have minimal impact on human health compared to pathogens. Gut microbiota was reported to be diverse in health and compositionally affected by ethnicity, geography, gender and pharmacological interventions. Perturbance in the composition of microbial communities has also been associated with metabolic disorders such as obesity, diabetes mellitus, insulin resistance, and immune and inflammatory disorders.(2)
The United States Human Microbiome Project sampled and sequenced the V3 to V5 regions in the 16S rRNA (ribosomal RNA) gene of 18 body sites. The study revealed that a number of taxa and gene families were differentially distributed with subject ethnicity at different body habitats.(3) Another study reported notable differences in bacterial species assemblages and functional gene repertoires between inhabitants of United States metropolitan areas and residents of Amazonas of Venezuela and rural Malawian communities.(4) In addition, among healthy young Chinese adults across nine provinces throughout China, species-level compositional profiles of the gut microbiome varied according to geography.(5) Chen et al recently reported greater species richness in the Caucasian population in the United States.(6)
Gender is another independent factor that is known to influence microbiome profile. It was seen to exert a statistically significant effect on human microbiome profiles, with a lower number of Faecalibacterium prausnitzii and higher number of Streptococci seen in women.(7) In the cohort from Rhode Island, New England, United States, Clostridium and Streptococcaceae were present in higher abundance in male and female subjects, respectively.(8) The Bacteroides-Prevotella group was reported to be in greater abundance in men than in women.(7,9) Dominianni et al found that gut microbiome in women was characterised by a lower abundance of Bacteroidetes.(10) In a recent study, gender was significantly associated with microbial beta diversity.(6) The gender-based differences may possibly be due to potential differences in gut physiology or a host effect on the microbiome.
Besides physiological factors, pharmacological interventions are known to alter the gut microbiome. Understanding alterations in human microbiome is important to identify individuals who are at risk of side effects from drug treatment such as antibiotics and proton pump inhibitors (PPIs). PPIs are commonly used for the treatment of acid-related disorders. The drug inhibits gastric acid secretion by blocking hydrogen/potassium (H+/K+) ATPases in gastric parietal cells, thereby increasing the intragastric pH.(11,12) Therefore, therapy with PPIs may perturb microbial communities and increase the risk of enteric infection and diarrhoea in humans.(13,14) Prolonged use of PPIs has been associated with stomach cancer risk and infections,(15) possibly due to modification of the normal flora by gastric acid inhibition. However, studies of PPIs and intestinal microflora interactions in the literature have focused primarily on Helicobacter pylori and Clostridium difficile, using culture-based techniques, and their relations with other bacteria were not well understood.
A few publications exploiting cutting-edge technologies and advanced bioinformatic tools to study the effect of PPIs on gut microbiome have recently emerged in the literature. Freedberg et al conducted a crossover study on healthy volunteers to examine if omeprazole altered gut microbiome.(16) Jackson et al also investigated the associations between gut microbiota composition and PPI use in a large healthy twin cohort.(17) Subsequent studies compared the faecal microbiomes of long-term PPI users to those with no history of PPI use in population-based studies, and examined if PPI use conferred a risk of Clostridium difficile infections.(18,19)
Currently, most microbiome studies have been conducted in Caucasian populations. To the best of our knowledge, this is the first study to interrogate microbiome composition in healthy individuals of both genders from the three major Asian ethnic groups (Chinese, Malay and Indian) and in the context of a clinically used therapeutic regimen of PPI (20 mg dose once daily for seven days) for common gastrointestinal conditions such as gastro-oesophageal reflux. A better understanding of the disease’s pathogenesis can lead to improved therapeutic strategies in applying microbiome interventions such as prescription of prebiotics, anti-microbial drugs and targeted faecal transplants.
METHODS
Healthy Chinese (n = 12; six male, six female), Malay (n = 12; six male, six female) and Indian (n = 10; four male, six female) subjects who were self-declared to be of the respective ancestry up to three generations participated in the study. Their mean age was 28.24 ± 4.38 (range 21–37) years.
Only subjects who had passed screening tests (liver and renal panel, full blood count) were enrolled. Two Indian subjects failed the screening and were excluded, one due to low haemoglobin level and the other due to urinary tract infection. The enrolled subjects provided a baseline stool sample (Day 1), took a course of PPI (omeprazole) at therapeutic dose (20 mg daily) for a duration of seven days (up to Day 7), and provided another stool sample seven days after cessation of the drug (Day 14). They were required to document any episodes of diarrhoea or side effects in their diary log. Included study subjects had not been on any antibiotic treatment, herbal supplements or probiotics/lactobacillus-containing products for at least one month prior to the study. All subjects provided written informed consent prior to their inclusion. The study was approved by the SingHealth Centralised Institutional Review Board (CIRB Ref 2014/2081), Singapore, and was performed in accordance with the ethical standards laid down in the Declaration of Helsinki (as revised in Brazil 2013).
The clinical study was conducted in the Clinical Trials and Research Unit, Changi General Hospital, Singapore. The study was designed to examine microbiome differences (a) among ethnic groups; (b) between the genders; and (c) before, immediately after and during the recovery period after stopping a course of seven-day omeprazole treatment. Omeprazole compliance was assessed by pill counting and measuring plasma drug concentrations using mass spectrometry. For comparison of ethnic and gender groups, only baseline (Day 1) samples were used.
Aliquots of fresh stool samples (approximately 3 g) that were collected in stool collection containers were immediately frozen and/or transported with ice packs to the laboratory within four hours of collection. The samples were stored at –80°C until DNA extraction. Microbial DNA was extracted from stool samples using the QIAamp DNA Stool Mini Kit (QIAGEN, Hilden, Germany).
The V3–V4 hypervariable region of the bacterial 16S rRNA gene was amplified in a solution with a total volume of 250 µL and containing 1× KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Cape Town, South Africa), 0.2 µM of each primer (V3/V4 Fwd: 5’-TCGTCGGCAGCGTCA GATGTGTATAA GAGA CAGCCTACGGGNGG CWGCAG-3’ and V3/V4 Rev: 5’-GTCTCGTGGGCTCGGAGATGTGTA TAAGAGACAGGACTAC HVGGGTATCTA ATCC-3’), and 12.5 ng of DNA. The polymerase chain reaction (PCR) conditions were: pre-denaturation at 95°C for three minutes, 25 cycles of denaturation at 95°C for 30 seconds, annealing at 55°C for 30 seconds and 72°C for 30 seconds, and a final extension at 72°C for five minutes. Successful amplification of the 1.5 kbp PCR product was verified using agarose gel electrophoresis. The PCR products were cleaned up using AMPure XP beads (Beckman Coulter, Brea, CA, USA). The purified amplified sequences were finely fragmented and libraries prepared using the Nextera XT Index kit (Illumina, San Diego, CA, USA). The resulting ‘end-adapter’ DNA fragment populations were cleaned up and validated using the Bioanalyzer DNA 1000 chip, followed by library quantification, normalisation and pooling. The pooled libraries were then denatured and subjected to 16S rRNA sequencing using the MiSeq Reagent Kit v2 (Illumina, San Diego, CA, USA) before they were loaded on the Illumina MiSeq platform. The 16S metagenomic data was captured on BaseSpace software (Illumina, San Diego, CA, USA). All experimental procedures, unless otherwise stated, were carried out according to the manufacturer’s instructions.
Filtering of sequences was performed in QIIME version 1.9.1 (Boulder, CO, USA) using the split_libraries.py and default settings as previously described.(20) High-quality reads were clustered into 97% operational taxonomic units (OTUs) via de novo uclust. Representative sequences were chosen from each OTU and assigned to taxonomic lineages with the RDP classifier using a confidence threshold of 80. Sørensen-Dice and Bray-Curtis dissimilarities for all pairwise comparisons of samples (beta diversity) as well as observed microbial species per sample (alpha diversity) were calculated in QIIME. Statistical analysis was performed using R packages.
Alpha diversity, which describes the richness and/or evenness of taxa within each group, was analysed using QIIME. The bioinformatics pipeline for performing microbiome analysis includes demultiplexing and quality filtering, OTU picking, taxonomic assignment, phylogenetic reconstruction, and diversity analyses and visualisations. Samples were examined using the alpha diversity indices of Simpson, Shannon, Chao1, phylogenetic diversity whole tree and observed species. Student’s t-test was used to compare the mean pairwise alpha diversity indices. A p-value < 0.05 was considered statistically significant.
Beta diversity compares the composition of the microbial communities to estimate the differences between comparison groups. Composition differences of microbial communities were studied using the Bray-Curtis dissimilarities and Sørensen-Dice dissimilarities function in the CRAN vegan package (
The CRAN vegan package was used to calculate the indicator value (IndVal) index, which is widely used in ecology for assessment of species-habitat association. IndVal was used to calculate both the occurrence and abundance of taxon. A high IndVal value (close to 1) indicates a strong association between the taxon (types of bacteria) and habitat (group of study). Taxa with lndVal value > 0.6 and p-value < 0.05 were selected as the signature taxon in each group from different taxonomic levels. Heatmaps were used to show composition differences of signature taxa between different study groups. A phylogenetic tree was constructed using all species or OTUs identified.
LefSe (Linear discriminant analysis [LDA] effect size;
RESULTS
After consuming omeprazole for seven days, subjects had an increase in species richness (p = 0.018) and a trend of increased alpha diversity (p = 0.14) that reverted to baseline on Day 14. No significant differences in alpha diversity indices were found among Chinese, Malays and Indians, and between male and female subjects. Box-whisker plots of the alpha diversity indices for treatment (Days 1, 7 and 14), ethnicity and gender groups are shown in
Fig. 1
Box-whisker plots show comparison of the alpha diversity indices (a) among Day 1, Day 7 and Day 14 treatment groups; (b) among Chinese, Malays and Indians; and (c) between male and female subjects using the Shannon index (left column) and Simpson index (right column). Student’s t-test was used to test the pairwise difference between groups.
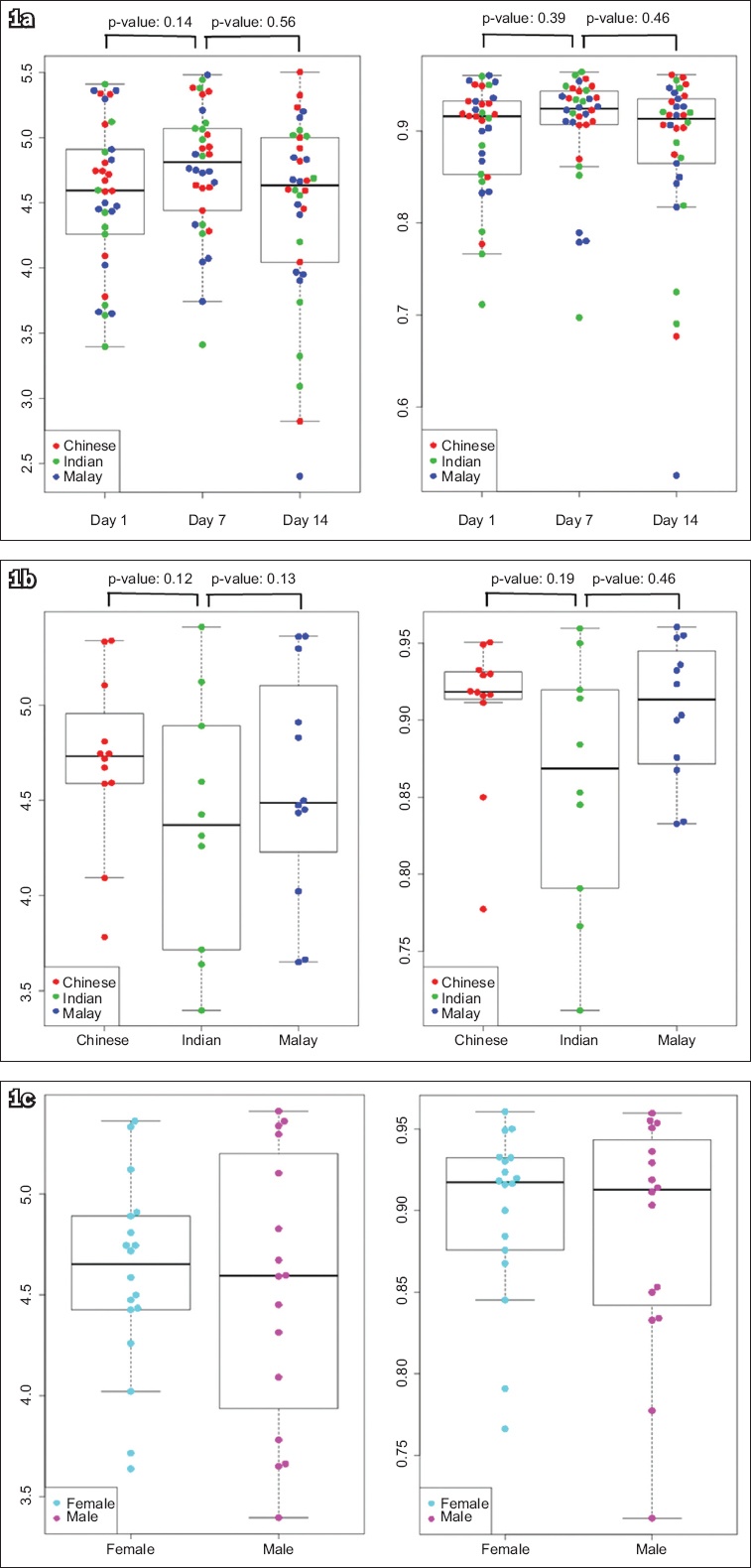
Table I
Alpha diversity indices from each study group.
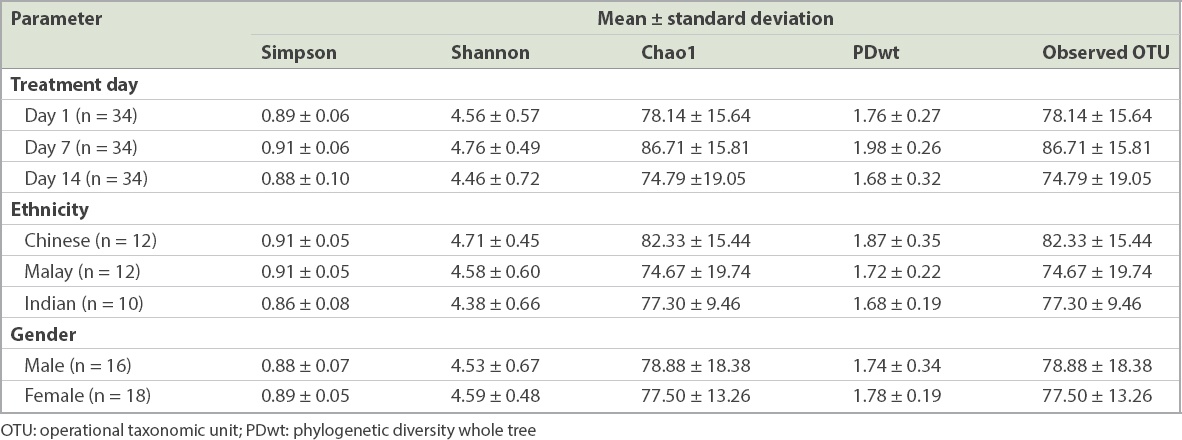
In terms of beta diversity, there was no apparent clustering of any subgroups indicative of compositional change at species, genus and family levels within the categories compared for PcoA plots based on both Bray-Curtis and Sørensen-Dice dissimilarities. The PcoA plots based on Bray-Curtis dissimilarities at species level based on treatment day, ethnicity and gender groups are shown in
Fig. 2
Principal coordinate analysis plots show Bray-Curtis dissimilarities at species level between microbiomes from different (a) treatment days; (b) ethnic groups; and (c) genders.
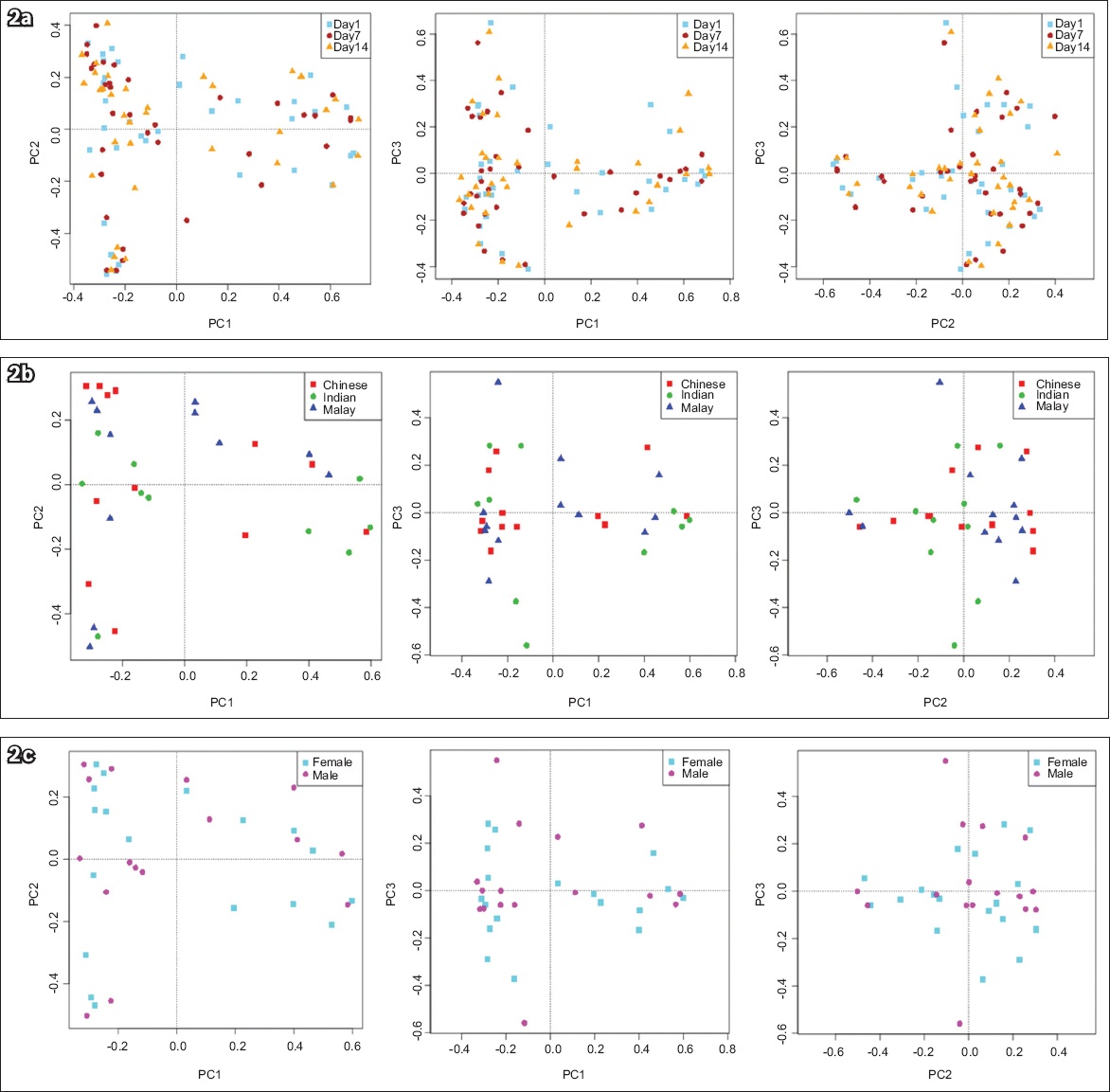
Comparing the groups according to treatment day, there was a significant increase in the relative abundance of the Bacilli class (p = 0.0001), Lactobacillales order (p = 0.0001), Streptococcaceae family (p = 0.0001), Streptococcus (p = 0.0001) and Veillonella genera, (p = 0.0041), and Streptococcus vestibularis (p = 0.0001) and Veillonella dispar (p = 0.0001) species immediately after PPI use (Day 7), which diminished seven days after stopping omeprazole on Day 14 (
Fig. 3
Graphs show the abundance of (a) Streptococcus vestibularis; and (b) Veillonella dispar on Days 1, 7 and 14.
Table II
Summary of signature taxa in Days 1, 7 and 14 treatment groups.
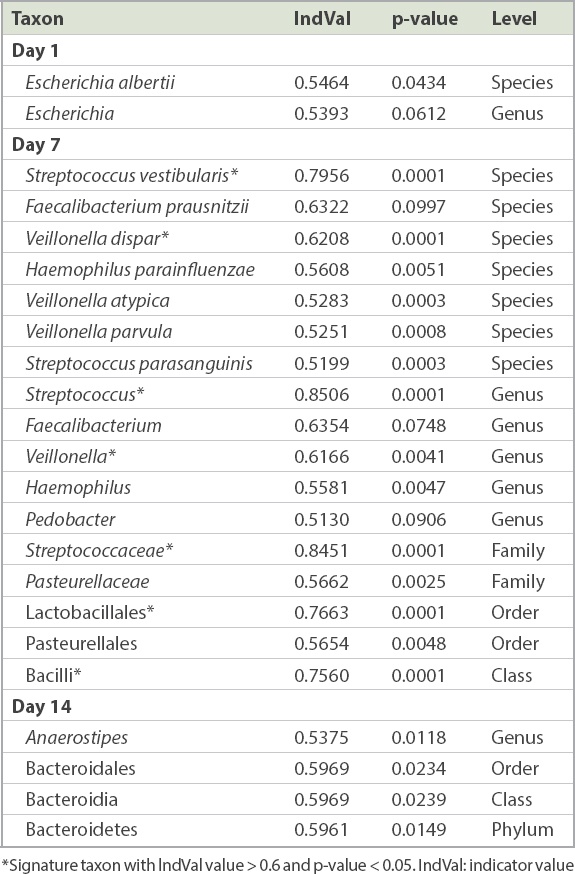
The presence of Faecalibacterium prausnitzii, Sutterella stercoricanis and Bacteroides denticanum was characteristic of Chinese, Malays and Indians, respectively (
Table III
Summary of signature taxa in Chinese, Indian and Malay subjects.
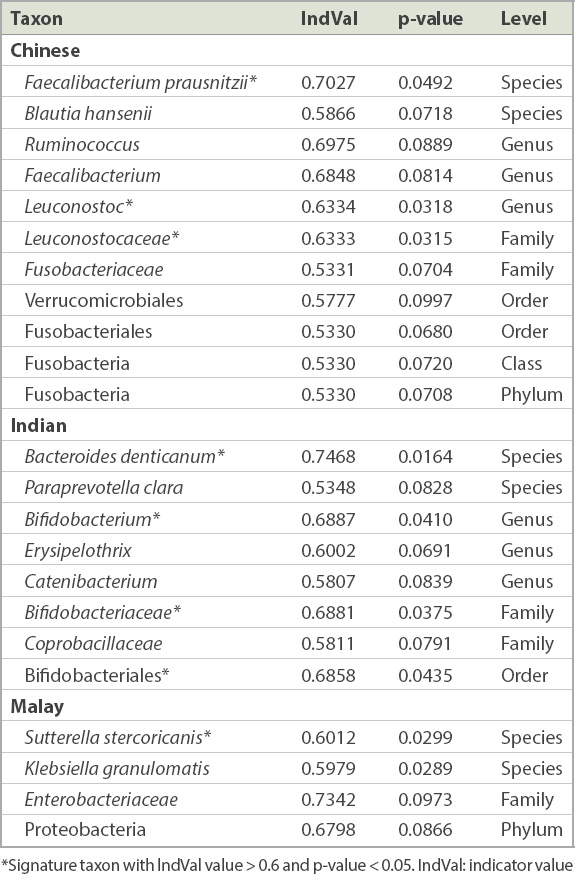
Table IV
Summary of signature taxa in male and female subjects.
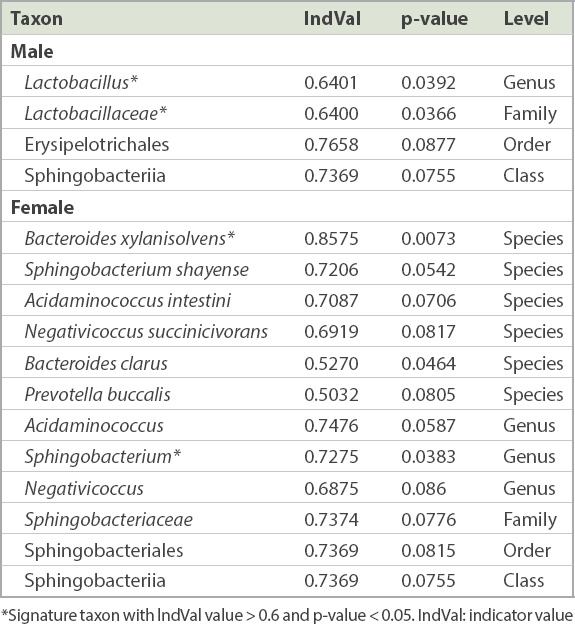
Fig. 4
Heatmaps show composition differences of signature taxa (a) among Day 1, Day 7 and Day 14 treatment groups; (b) among Chinese, Malays and Indians; and (c) between male and female subjects using Z-score transformed relative abundance data.
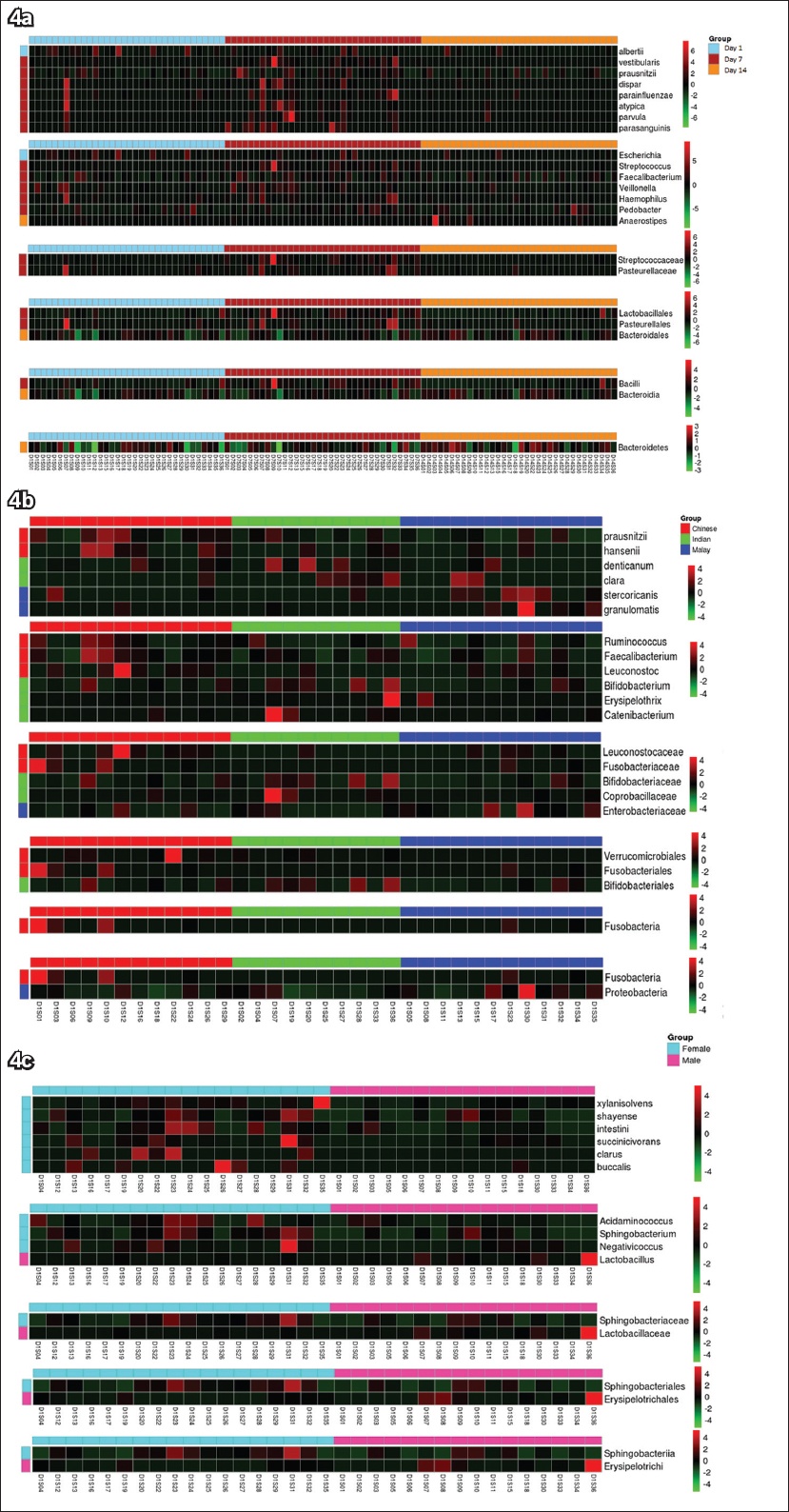
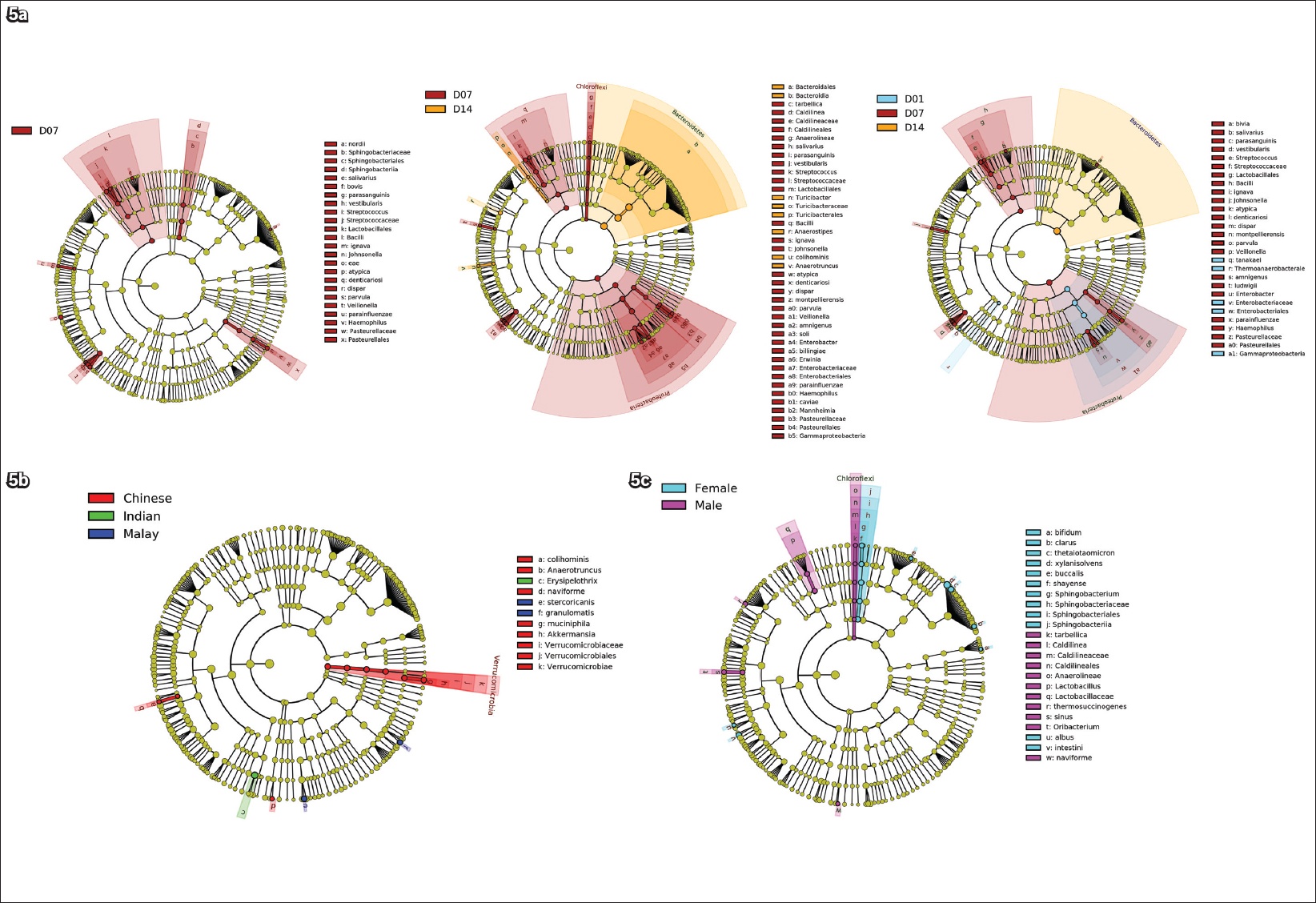
Fig. 5
Cladograms show taxonomic representations of statistically and biologically consistent differences (a) among Days 1, 7 and 14 treatment groups; (b) among Chinese, Malays and Indians; and (c) between male and female subjects. The diameter of each circle is proportional to the taxon’s abundance.
DISCUSSION
This study evaluated the baseline gut microbiome pattern of a cohort of 34 Asian subjects that was almost equally distributed in terms of gender and ethnicity, as it comprised the three major ethnic groups of the local population, Chinese, Malays and Indians. The effect of omeprazole on gut microbiome on Day 7 and Day 14 was examined. The parameters analysed were alpha diversity index, beta diversity index, signature taxa identification and LDA effect size.
We found that seven days of omeprazole treatment (20 mg dose once daily) acutely increased faecal microbiome alpha diversity and species richness, although the effect reverted to baseline after seven days of omeprazole cessation (
Signature taxa identification revealed a strong association between the relative abundance of the Bacilli class, Lactobacillales order, Streptococcaceae family, Streptococcus and Veillonella genera, and Streptococcus vestibularis and Veillonella dispar species, and omeprazole usage. These findings are consistent with the report by Imhann et al, which associated the increase of both genera with PPI use.(19) Veillonella was found to form co-aggregate complexes with Streptococcus and positively correlate to the abundance of Streptococcus in irritable bowel syndrome (IBS) caused by small intestinal bacterial overgrowth.(21,22) This positive association was also evident in the present study, encouraging speculation about the possible risk of developing IBS with PPI use. Antharam et al also found that Veillonella and Lactobacillus were enhanced in patients with Clostridium difficile infections compared with healthy controls.(23) Among the taxa, Streptococcus is most well recognised as an organism that has been shown, across several studies, to increase the risk of Clostridium difficile infections.(17-19,23) Both Streptococcus vestibularis and Veillonella dispar species belong to the Firmicutes phylum, are gram-negative bacteria and inhabit the human oral cavity.(24,25) Interestingly, all the species that emerged as signature taxa on Day 7 with statistical significance (
The taxa Proteobacteria phylum, Gammaproteobacteria class, Enterobacteriales order and Enterobacteriaceae family, which were only associated with the Day 7 group when it was compared to the Day 14 group, but not when it was compared to the Day 1 group, showed a gradual reduction from Day 7 with a significant drop by Day 14 although it is usually present at higher levels in baseline faecal sample. The negative association of Enterobacteriaceae with omeprazole use is in contrast to other reported studies(16,19) and is likely a transient phenomenon of the short treatment regimen. The association of Bacteroidetes phylum with the Day 14 group when compared to the Day 1 and Day 7 groups suggests the delayed effect of omeprazole treatment. However, this finding is inconclusive and warrants further research to shed more light on the mechanism.
Faecalibacterium prausnitzii, Sutterella stercoricanis and Bacteroides denticanum were characteristic of Chinese, Malays and Indians, respectively. Faecalibacterium prausnitzii is a commensal bacterium of the human gut flora that synthesises butyrate and other short-chain fatty acids through the fermentation of dietary fibre.(27) As butyrate is an essential energy source for colonocytes,(28) the presence of colonic butyrate is known to inhibit the growth of Clostridium difficile.(29) This may explain the higher prevalence of Clostridium difficile infections among white patients as compared to Asian patients.(30) Faecalibacterium prausnitzii is also known for its anti-inflammatory role and providing protection against Crohn’s disease.(31) The roles of Sutterella stercoricanis and Bacteroides denticanum in humans are largely unknown, except for one report that linked the former to gastrointestinal disturbances in autistic children.(32) The inter-ethnic microbiome differences could be due to the different dietary habits among ethnic groups. Lactobacillaceae, a family of lactic acid bacteria and Bacteroides xylanisolvens, a commensal strain, were the signature taxa of male and female subjects, respectively. Bacteroides xylanisolvens was believed to confer probiotic health benefits,(33) but has recently been associated with gout.(34) None of the taxa that distinguish ethnicity and gender overlapped with the signature taxa of omeprazole treatment.
We acknowledge that this study had several limitations. The treatment period of seven days was relatively short as compared to other studies. Therefore, the gut microbiome might not have stabilised and it was possible that the bacteria was still in the process of adapting to the new environment. Nevertheless, the findings offer an insight into the short-term or interim changes in gut microbiome, with possible long-term implications. Also, the sample size was relatively small and the study was therefore not powered to detect subtle changes in microbial diversity. In addition, baseline microbiome variability was not determined, as we did not sample multiple faecal samples prior to the initiation of omeprazole treatment. Lastly, the microbiome profiling provided no indication of the actual functionality and health effects on the individual. Further functional and clinical studies are required to elucidate the mechanistic pathophysiology associated with microbiome alterations due to PPI usage.
In spite of these limitations, to the best of our knowledge, this is the first study to: establish the baseline human gut microbiome profile in healthy individuals from the three major ethnic groups of the Asian population, who were equally represented in terms of gender; and examine the effect of a commonly used therapeutic regimen of omeprazole on gut microbiome. The knowledge gained from this research paves the way for future studies to investigate the relationship between gut microbiome and various disease states such as obesity and inflammatory conditions, and for the implementation of microbiome intervention strategies such as probiotic or prebiotic therapy and faecal transplants in clinical practice. It can also aid in the development of PPI prescription guidelines and policies for medical professionals, to avoid overutilisation of PPIs and the consequent risk of enteric infections.
ACKNOWLEDGEMENTS
The study was financially supported by a research grant (CHF2014.03-P) from Changi General Hospital, Singapore, awarded to Ang TL. We would like to thank all staff of the Clinical Trials and Research Unit for their assistance in conducting the trial.