

Abstract
Ellis-van Creveld (EvC) syndrome is a rare autosomal recessive malformation disorder. Cardiac defects are observed in about 50% of EvC cases. Surgical data is lacking on the prognosis and life expectancy of EvC patients. Herein, we report the case of a 38-year-old man with EvC syndrome who underwent two surgical corrections for cardiac anomalies. This report supplements the available information on the clinical course of EvC syndrome in older patients.
INTRODUCTION
Ellis-van Creveld (EvC) syndrome is a rare autosomal recessive malformation disorder. Congenital cardiovascular malformations, disproportionate limb dwarfism, postaxial polydactyly and ectodermal dysplasia are all clinical features of EvC syndrome.(1) EvC syndrome is caused by an anomaly of chromosome 4p16.2.(2) In 50%–60% of cases, congenital heart defects are present, with single atrium being the most common cardiac malformation.(2,3) Data is lacking on the specific surgical prognosis and postoperative life expectancy of patients with rare conotruncal defects such as EvC syndrome.(4) We herein present the case of a 38-year-old man with EvC syndrome who underwent two surgical corrections for cardiac anomalies during adulthood. The management and perioperative care of the patient are also described.
CASE REPORT
The patient was a 38-year-old Caucasian man of Spanish origin. His height was 165 cm and his parents were cousins. Both parents were of normal height and neither showed any minor anomalies of the EvC syndrome spectrum. The patient had two healthy sisters. His morphological features were: (a) normal mouth opening with missing lower incisors (
Fig. 1
Photograph shows a normal mouth opening and missing lower incisors on the patient.
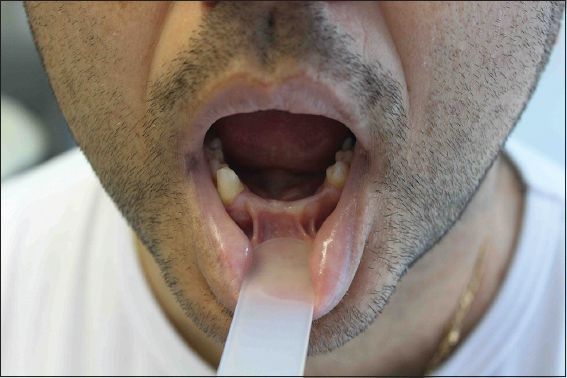
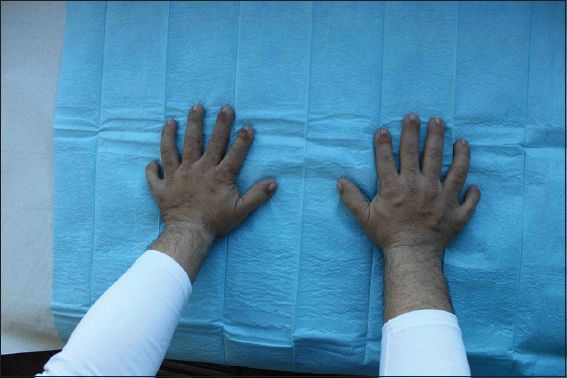
Fig. 2
Photograph shows the patient’s disproportionately short extremities and additional postaxial digit on each hand.
In 1998, the patient underwent successful ostium primum atrial septal defect closure through a midsternotomy. He re-presented with embolic cerebral ictus in 2007, and cavotricuspid isthmus ablation for common atrial flutter was performed successfully. There were no serious aftereffects.
During a routine examination in 2012, a high-pitched systolic murmur was discovered. Electrocardiography showed sinus rhythm with hemiblock and right bundle branch block, while echocardiography revealed a dilated and hypertrophic left ventricle. These abnormalities produced a flow that was directed to the left atrial appendage, leading to severe mitral valve regurgitation. The patient’s systolic pulmonary artery pressure was 50 mmHg and his aortic valve was normal. His medical treatment included ramipril, duloxetine, alprazolam and acenocumarol. The patient was scheduled for mitral valve surgery.
In the operating theatre, the patient’s right femoral artery was cannulated. A right thoracotomy was performed on the fourth intercostal space and bicaval cannulation was established. Both veins were excluded. The operation used a normothermic cardiopulmonary bypass without cross-clamping of the aorta in the beating heart. To avoid air embolisms, continuous carbon dioxide (CO2) field flooding was applied using a CarbonAid CO2 diffuser (Cardia Innovation AB, Stockholm, Sweden). The attempt to perform a mitral valve repair was unsuccessful and a bileaflet mechanical valve (no. 29) was implanted. No complications associated with anaesthesia occurred. The patient’s postoperative recovery was uneventful and he was discharged on postoperative day 10. After two years of follow-up, echocardiography revealed optimal function of the mitral valve and a decrease in systolic pulmonary artery pressure (30 mmHg).
DISCUSSION
EvC syndrome is a rare disease that was first described by Ellis and van Creveld in 1940.(6) It is a chondral and ectodermal dysplasia characterised by short ribs, polydactyly and ectodermal and heart defects,(1,3) and is inherited as an autosomal recessive disease.(3)
The three most common cardiac defects in patients with EvC are atrioventricular canal defect, common atrium and persistent left superior vena cava. Other cardiac defects include transposition of the great arteries, pulmonary stenosis, anomalous pulmonary venous return, tricuspid atresia and left-sided obstructive lesions.(1-3) Almost half of all EvC patients die during childhood due to cardiorespiratory complications and only a few have undergone open heart surgery.(3) Perioperative morbidity may result from difficulties with airway management due to cleft lip/palate, dental/oral malformations, fusion between the inner lip and gum, and maxillary/mandibular deformities.(1)
In a recent study, O’Connor and Collins analysed the surgical outcomes of 32 children with EvC syndrome and reported a high mortality rate of 44%, most of which were related to respiratory complications. The children who survived the surgery experienced significantly more postoperative respiratory morbidity.(7)
Most studies on the surgical prognosis and life expectancy of EvC patients after cardiac surgery were centred on paediatric patients.(2,7) Compared to the present case, most of the EvC patients reported in the literature had shorter follow-up (e.g. 30 days).(7) In EvC patients who are of advanced age, there is still a paucity of information on surgical prognosis and life expectancy.(8,9) Baujat et al reported the case of a healthy 21-year-old woman with dental morphologic anomalies who was diagnosed at birth with EvC syndrome without heart involvement.(10) Rudnik-Schöneborn et al highlighted the clinical course of EvC syndrome with increasing age in their report of two patients with skeletal malformations that were diagnosed in adulthood (at 18 years and 30 years, respectively). These patients underwent corrective surgery and had proper follow-up. The second patient had previously undergone corrective surgeries for common atrium (at the age of eight and 12 years).(8) The patient reported herein was a 38-year-old man whose health-related quality of life was good after two heart operations. In our review, we observed that multidisciplinary therapeutic planning is the cornerstone of EvC syndrome management. We also discovered that the prognosis of EvC syndrome depends on whether breathing difficulties were present during the first months of life.(10) Skeletal malformations are the predominant and disabling feature that should be promptly treated in order to avoid permanent disability.
In conclusion, this case report, which described an EvC patient who survived longer than most other patients with similar conditions, adds important clinical information on the long-term course of EvC syndrome. Our case also shows that EvC patients can reach adulthood and have the potential to attain a relatively good quality of life.