Author Information >Copyright and License information >
1College of Life Sciences and Medicine, Zhejiang Sci-Tech University, Hangzhou, China 2College Hospital, Zhejiang Sci-Tech University, Hangzhou, China 3Shanghai Yuyan Cell Research Company, Shanghai, China 4Johnson and Johnson Pharmaceutical Research and Development LLC, San Diego, CA, USA Correspondence: Dr Jian Chen, Associate Professor, College of Life Sciences and Medicine, Zhejiang Sci-Tech University, Hangzhou 310018, Zhejiang, China. chj1999@126.com
Overproduction of immunoglobulin E (IgE) by a subset of B cells plays a key role in the pathogenesis of allergic asthma. Anti-IgE monoclonal antibodies have been successfully used to treat the disease, but long-term application is required.
METHODS
For this study, cytotoxic T lymphocytes (CTLs) against IgE-producing B cells were generated ex vivo by stimulating naive CD8 T cells with IgE-derived peptides presented by Drosophila-derived artificial antigen-presenting cells. Based on the treatment of allergic asthma in mice, the inhibitive effect of this CTL on IgE responses and airway inflammation was determined with the enzyme-linked immunosorbent assay and histochemical method.
RESULTS
The IgE-specific CTLs effectively lysed target cells in vitro, while the adoptively transferred CTLs specifically inhibited IgE responses and airway inflammation in an asthmatic mouse model. The effect of IgE-specific CTLs is MHC (major histocompatibility complex) Class I-restricted and requires the expression of perforin.
CONCLUSION
IgE-specific CTLs generated ex vivo may provide a novel treatment for allergic asthma and lead to a new therapy for other immunological disorders.
Keywords: artificial antigen-presenting cells, cytotoxic T lymphocytes, immunoglobulin E, perforin
INTRODUCTION
Elevation of serum immunoglobulin E (IgE) level is correlated with atopic diseases such as allergic rhinitis and allergic asthma.(1) Circulated IgE in the blood binds to the high-affinity IgE receptor (FcεRI) on the surface of mast cells positioned along the mucosal lining and underneath the skin during an allergic response. IgE-mediated mast cell degranulation also results in the release of newly synthesised inflammatory cytokines and chemokines, including IL-4, IL-5, IL-6, TNF, MCP-1 and MIP-1, and thus plays a role in the late phase of allergic responses.(2) Therefore, IgE is usually used as a natural target for anti-allergic therapy. Indeed, anti-IgE monoclonal antibodies (mAbs) have successfully been developed for the treatment of IgE-mediated allergic disorders.(3-7) It has been demonstrated that anti-IgE mAbs attenuate both early- and late-phase asthmatic responses, improving both the symptom score and peak expiratory flow in patients with allergic asthma.(8-10) To benefit from this treatment, however, patients required the injection of a high dose of anti-IgE antibody (150–300 mg/injection) every 2–4 weeks.(11) Despite a near-complete suppression of free IgE, a prolonged and substantial rise in total IgE, in the form of IgE-anti-IgE complexes, was observed in patients who received anti-IgE antibody,(3) suggesting that IgE synthesis could not be stopped by the treatment.(9,12,13) Thus, the direct inhibition of IgE-producing cells by IgE-specific cytotoxic T lymphocytes (CTLs) may be an alternative approach to the long-term treatment of allergic disorders.
CTLs play an essential role in immunity against viral and intracellular pathogens by recognising antigenic peptides in the context of major histocompatibility complex (MHC) Class I molecules.(14) Previously, we showed that Drosophila-derived artificial antigen-presenting cells (aAPC) can be used to generate CTLs specific for tumour-associated antigenic peptides in vitro, and that the adoptive transfer of these CTLs inhibits tumour cell growth in vivo.(15) Therefore, we hypothesised that adoptive transfer of these IgE-specific CTLs should also be able to lyse IgE-producing B cells in vivo and consequently lead to inhibition of IgE-mediated pathology. This study aimed to evaluate whether IgE antigenic peptide-specific CTLs can be generated in vitro and to test if these CTLs could inhibit IgE responses in vivo. Using an established aAPC system,(16) we successfully generated IgE-specific CTLs in vitro, the adoptive transfer of which efficiently inhibited IgE responses and airway inflammation in an asthmatic mouse model.
METHODS
C57BL/6J (B6), CB6F1/J (BALB/cJ F × C57BL/6J M), C57BL/6-Pfp (perforin-/- [PF-/-]), B6.129S7-Ifng (interferon gamma-/- [IFN-γ-/-]) and B6.129P2-b2m (β-2-microglobulin-/- [β2m-/-]) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and raised at room temperature in an individually ventilated cage system with conventional conditions. All experimental animals used in this study were under a protocol approved by the Institutional Animal Care and Use Committee.
All IgE-derived peptides were synthesised by SynPep Corporation (San Diego, CA, USA) and purified with C18 reverse phase high-performance liquid chromatography. RMA-S cells and Ld-transfected RMA-S cells (RMA-S.Ld) were cultured and maintained as previously described.(16) Drosophila cells transfected with MHC Class I (Ld or Db), CD54 (also ICAM-1) and CD80 (also B7-1) were generated and maintained as previously described.(16) The MHC Class I stabilisation assay was applied, also as previously described.(17) During the assay, RMA-S or RMA-S.Ld cells were cultured overnight at room temperature with or without indicated peptides and then shifted to an additional two-hour culture at 37°C. The cells were stained with fluorescein isothiocyanate-labelled anti-H-2 Db or Ld mAb (BD Pharmingen, San Diego, CA, USA) and were analysed with a FACScan flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
CD8 T cells were purified from lymph nodes of CB6F1/J, B6/J, IFN-g or PF-deficient mice (IFN-g-/- or PF-/-) using a positive selection magnetic-activated cell sorting system (Miltenyi Biotec, Somerville, MA, USA). Purified CD8 T cells were cultured with Drosophila cells transfected with Db, CD54 and CD80 with IgE 44 peptide (IgEp44) or IgE 366 peptide (IgEp366). IL-2 (20 hU/mL) was added into the culture medium at Days 3, 5 and 7, and cells were split on Day 5 and Day 7 based on cell density. CTLs were harvested at Day 9, and their specific activity was tested as previously described.(16) RMA-S or RMA-S.Ld cells were incubated with 51Cr for one hour with or without IgE peptides and were used as target cells. The CTL effector cells were cultured with the labelled target cells at indicated effector-to-target ratios in a U-bottom microplate at 37ºC for four hours. Specific 51Cr release was calculated as previously described.(16)
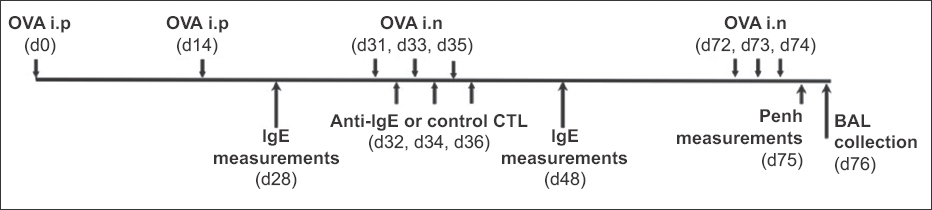
The mouse model of allergic asthma was generated as indicated in Fig. 1.(18) CB6F1/J, C57B6/J or β2m-/- mice were pre-sensitised using intraperitoneal injections of alum-precipitated ovalbumin (OVA; 100 mg) on Day 0 and Day 14, respectively. The mice were then challenged with 1 mg of OVA (20 µL) on Days 31, 33 and 35, and anti-IgE or control CTLs (5–10 × 106/mouse) were given intravenously between the challenges (Days 32, 34 and 36). Two weeks after CTL treatment, serum was collected from each mouse and the total IgE or OVA-specific IgE was measured with enzyme-linked immunosorbent assay (ELISA). Five weeks after CTL treatment, the mice were intranasally re-challenged with equal amounts of OVA every day for three days (Days 72, 73 and 74). At Day 75, airway responsiveness to increased doses of methacholine was assessed using a whole body plethysmography system (BUXCO Electronics, Troy, NY, USA). Each methacholine dose was given over a five-minute period, and the maximal enhanced pause (Penh) value was measured during the following five-minute period. On Day 76, the mice were sacrificed, and bronchoalveolar lavage (BAL) and lung tissues were prepared from each mouse. To analyse treating effects, a series of assays were performed, including cell counting, haematoxylin and eosin (H&E) staining for differentiating eosinophils, and ELISA for measuring eotaxin in BAL. For the long-term study of anti-IgE CTLs in vivo, the pre-sensitised and treated mice were re-challenged at two, four and six months after CTL therapy. Total serum IgE level was measured one week after each re-challenge.
Fig. 1
Diagram shows the experimental design of a mouse model of allergic asthma. BAL: bronchoalveolar lavage; CTL: cytotoxic T cell; d: Day; IgE: immunoglobulin E; i.n: intranasal; i.p: intraperitoneal; OVA: ovalbumin
Total serum IgE and immunoglobulin G (IgG) isotypes (IgG1, IgG2a) and eotaxin from the mice were determined by sandwich ELISA. For this purpose, polystyrene 96-well plates (Costar, Corning, NY, USA) were coated overnight at 4°C with capture antibody of anti-mouse IgE (BD Pharmingen, San Diego, CA, USA) or anti-IgG (Southern Biotechnology, Birmingham, AL, USA) or anti-eotaxin (R&D, Minneapolis, MN, USA) in phosphate-buffered saline (PBS; 100 µL/well). Plates were then washed with PBS plus 0.02% Tween-20 and blocked with 2% bovine serum albumin (100 µL/well) for two hours at room temperature. After washing and blocking, diluted serum samples and standards were added to each well and incubated at room temperature for two hours. For measurement of total IgE, the plates were washed and incubated with biotinylated anti-mouse IgE for one hour, followed by incubation with horseradish peroxidase-conjugated streptavidin for an additional hour. The plates were washed and developed with 3,3’,5,5’-tetramethylbenzidine solution (BD Pharmingen, San Diego, CA, USA). For measurement of IgG1 and IgG2a, the plates were washed and incubated with alkaline phosphatase-conjugated rabbit anti-mouse IgG1 or IgG2a for one hour. After washing, the plates were developed with para-nitrophenyl phosphate substrate. For measurement of OVA-specific IgE or IgG, the determination procedure was carried out as described herein, but the plates were coated with 100 µL of OVA (10 µL/mL) instead of capture antibody. Absorbance was measured with a microplate reader at 450 nm wavelength.
GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA) was used for statistical analysis. Mean methacholine dose-response curves for each experimental group were compared using two-way analysis of variance. All data was presented as mean ± standard deviation. Student’s t-test was used for all comparisons. A p-value < 0.05 or < 0.01 was considered statistically significant or extremely significant, respectively.
RESULTS
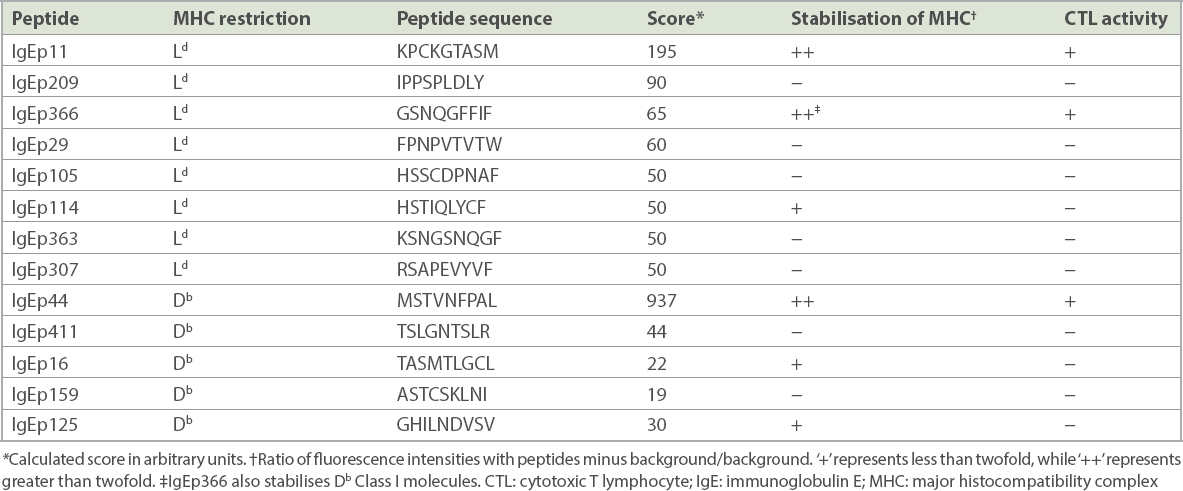
The amino acid sequence of the constant region of IgE was analysed for 9-mer peptide sequences that contained binding motifs for H-2 Ld and Db MHC Class I molecules by using the BIMAS program of the Bioinformatics and Molecular Analysis Section (https://www-bimas.cit.nih.gov/molbio/hla_bind/). This program ranks potential nonapeptides based on a predicted half-life of dissociation to MHC Class I molecules.(19) Based on the ranking analysis, eight nonapeptides with Ld binding motifs and five nonapeptides with Db binding motifs were selected for synthesis and further analysis (Table I).
Table I
Identification of antigenic peptides for mouse IgE.
We tested the capacity of these synthetic peptides to bind and stabilise Ld and Db Class I molecules expressed on Drosophila (Schneider 2) cells and RMA-S cells.(17,20,21) Two IgE-derived peptides, identified as IgEp11 and IgEp366, bound strongly to Ld, whereas IgEp114 bound only weakly to Ld. Of the five peptides predicted to bind to Db, only IgEp44 bound strongly, while IgEp16 and IgEp125 bound weakly to Db. Interestingly, IgEp366, which was originally predicted to bind Ld, bound Db as well. Therefore, a total of six nonapeptides were identified that bound to either Ld or Db MHC Class I molecules (Table I).
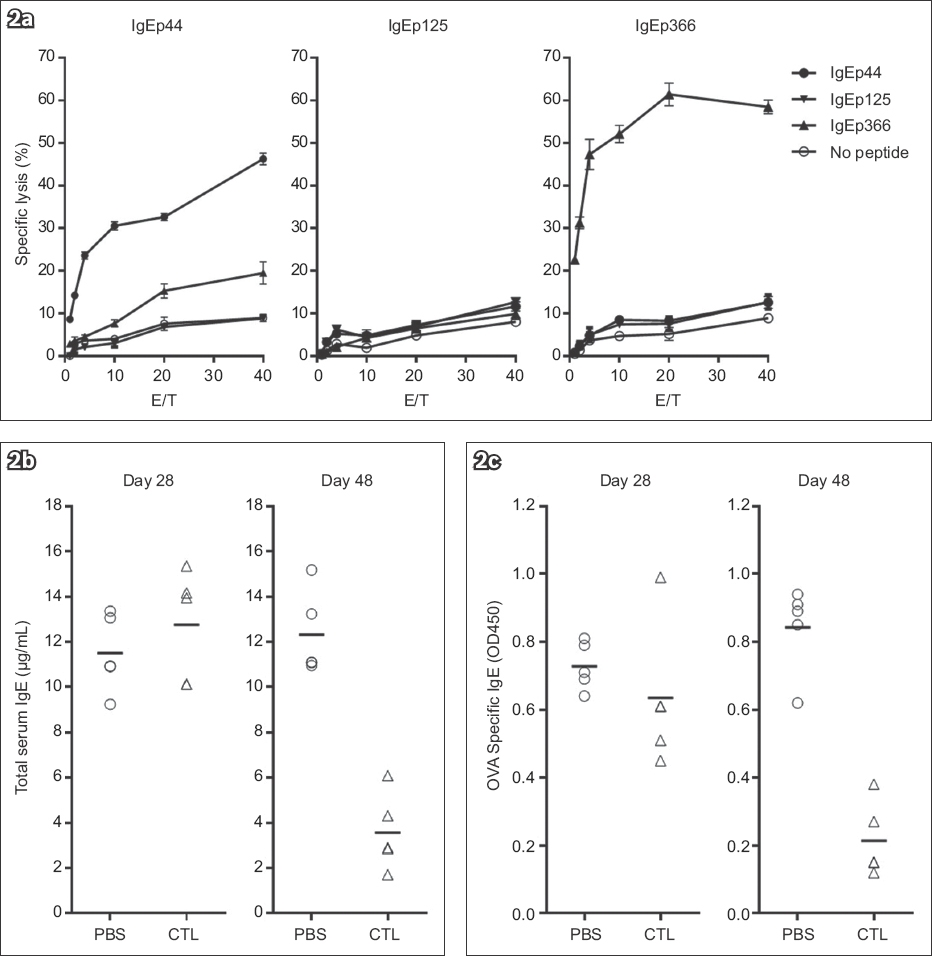
The ability of the IgE-derived peptides to elicit CTL responses was initially evaluated in vitro. As previously described,(16) Drosophila cells transfected with MHC Class I, CD54 and CD80 molecules are potent antigen-presenting cells (APC) for the activation of resting naive CD8 T cells in vitro. Resting naive CD8 T cells purified from mouse lymph nodes were cultured with transformed Drosophila cells expressing Ld or Db, CD54 and CD80 (Fly Ld.B7.ICAM or Fly Dd.B7.ICAM) in the presence of indicated peptides, and the CTL activity was measured on Day 9. The IgEp44-induced CTLs specifically lysed RMA-S cells loaded with the corresponding peptide. The target cells alone or those loaded with control IgE peptides were not recognised by IgEp44-specific CTLs (Fig. 2a). As expected, IgEp366 bound both Ld and Db, and induced Ld-restricted CTLs (data not shown) as well as Db-restricted CTLs. Although IgEp16 and IgEp125 were demonstrated to bind Db, no specific CTL activity was raised by these two peptides (Fig. 2a and Table I). Because CTLs induced by IgE-derived peptides specifically killed target cells in vitro, we were interested in determining if these CTLs were capable of inhibiting IgE responses in vivo. As spontaneous allergic responses were lacking, an experimental mouse model was developed to evaluate the effect of IgE-specific CTLs in vivo.(18,22) To evaluate both Db and Ld restricted peptides, CB6F1/J mice were used for the experiments (Fig. 1). In the initial experiments, we administered IgE-specific CTLs during immunisations with OVA and found that the levels of both total serum IgE and OVA-specific IgE were dramatically reduced in the mice that received IgE-specific CTLs (data not shown). To closely mimic the clinical situation of asthmatic patients, who are already sensitised to allergens when treatment is initiated, we challenged pre-sensitised mice with OVA intranasally and administered the IgE-specific CTLs. Intranasal challenge with the antigen induced further elevation of serum IgE in the pre-sensitised control mice (Figs. 2b & c). However, the responses by both total serum IgE and OVA-specific IgE were markedly reduced in the CTL-treated mice. Thus, adoptive transfer of IgE-specific CTLs during either immunisation or a re-challenge inhibited IgE responses in the mouse model of allergic asthma.
Fig. 2
Charts show (a) the generation and target specificity of anti-immunoglobulin E (IgE)-specific cytotoxic T lymphocytes (CTLs) in vitro; and (b) total serum IgE response and (c) antigen-specific IgE response before (Day 28) and after CTL treatment (Day 48), demonstrating the inhibition of IgE response by specific CTLs. CD8 T cells were purified from lymph nodes of CB6F1/J mice and stimulated by co-culturing with Drosophila-derived artificial antigen-presenting cells expressing H-2 Db, CD54 and CD80 molecules in the presence of indicated IgE peptide. CTL activity was measured with induced CD8 T cells against 51Cr labelled RMA-S cells in the presence of various IgE peptides. After the second immunisation, each mouse received 1 × 107 IgE-specific CTLs, except for the control mice, which received phosphate-buffered saline (PBS). There were five mice in each CTL or PBS group. E/T: effector-to-target ratio; OVA: ovalbumin
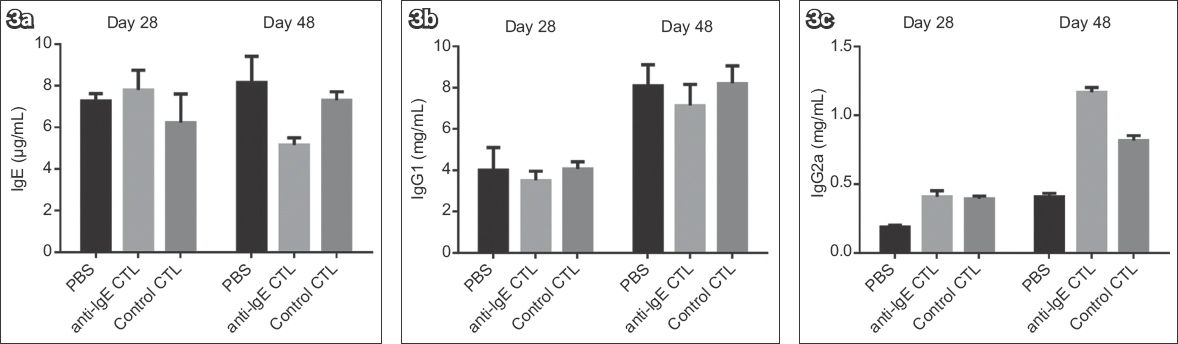
To determine if IgE-specific CTLs specifically inhibit IgE responses exclusively, both IgE and IgG responses were investigated (Fig. 3). Although IgE-specific CTLs effectively inhibited IgE responses in vivo (Fig. 3a), neither IgG1 nor IgG2a responses were inhibited by the cells (Figs. 3b & c). In contrast, IgG2a responses were enhanced by this treatment. To investigate whether the inhibition of IgE response was a consequence of the specific nature of the IgE CTLs, we generated CTLs specific for a Db-restricted peptide NP366 derived from influenza A virus nuclear protein and administered it as a specificity control. The control CTLs specifically lysed target cells loaded with NP366 peptide (data not shown), whereas IgE responses were unaffected. Similarly, IgG2a responses were enhanced by the control CTLs (Fig. 3c) as well as by the IgE-specific CTLs. ELISA determination also indicated that IgG1 and IgG2a were not OVA-specific (data not shown). Therefore, we concluded that IgE-specific CTLs specifically inhibit IgE responses and indirectly enhance IgG2a responses.
Fig. 3
Charts show the effect of anti-immunoglobulin E (IgE) cytotoxic T lymphocytes (CTLs) on the responses of (a) IgE, (b) IgG1 and (c) IgG2a, measured before CTL (Day 28) and after CTL treatment (Day 48). Anti-IgE CTLs were generated as described in Fig. 1, and control CTLs were generated with an influenza nuclear protein antigenic peptide. Two weeks after the second immunisation, the mice were challenged with ovalbumin intranasally every other day, for a total of three times. IgE-specific or control CTLs (5 × 106) were given intravenously one day after each challenge. There were five mice in each group. PBS: phosphate-buffered saline.
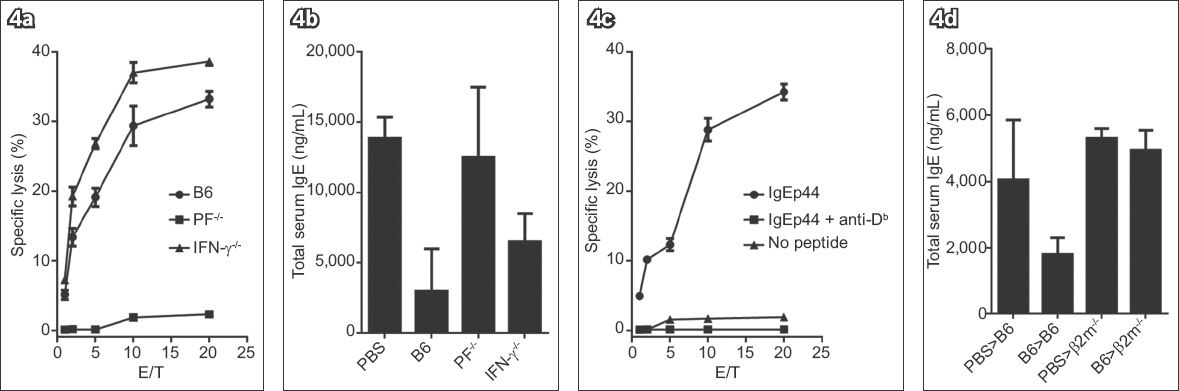
CTL cytotoxic activity primarily depends on the expression of PF alone. In addition, CTLs also produce some other cytokines, such as IFN-γ, to regulate Th1 response. It was therefore possible that the inhibition of IgE responses by CTLs was a consequence of regulation of Th1 responses by CTL-produced IFN-γ than a direct killing result of IgE-producing cells.(8) To distinguish the influence of the killing activity and IFN-γ release on IgE responses, we generated IgE-specific CTLs from either PF-/- or IFN-γ-/- mice.(23,24) CD8 T cells purified from PF-/- mice proliferated in response to IgE peptides. However, these PF-/- mice-derived CTLs were unable to kill target cells loaded with IgE peptides and therefore lost the ability to inhibit IgE responses in vivo (Figs. 4a & b). Contrarily, the IFN-γ-/- mice-derived CTLs specifically killed the target cells (Fig. 4a) and showed a similar inhibition effect on IgE responses in vivo to that of wild type (WT) CTLs (Fig. 4b). These results suggested that the expression of PF, rather than IFN-γ, was required for CTL activity, and that the validated inhibition of IgE response by IgE-specific CTLs most likely resulted from PF-mediated killing of IgE-producing cells.
Fig. 4
Charts show the effect of perforin (PF) or interferon gamma (IFN-g) expression on (a) anti-immunoglobulin E (IgE) cytotoxic T lymphocyte (CTL) activity and (b) inhibition of IgE responses in vivo. IgE-specific CTLs were generated from either wild-type (WT) or knockout strains of mice (PF-/- or IFN-γ-/-). CTL activity was evaluated as described in Fig. 2. Chart shows (c) results of CTL assay, performed in the absence or presence of anti-major histocompatibility complex (MHC) Class I (Db) antibody, with IgE-specific CTLs generated from WT mice; and (d) requirement of MHC Class I expression on anti-IgE CTLs in vivo in pre-sensitised (B6 or b2m-/-) mice. Total serum IgE was measured two weeks after treatment with anti-IgE CTLs. For the in vivo experiments, there were five mice in each group. E/T: effector-to-target ratio; PBS: phosphate-buffered saline
CTLs, as effector cells, recognise peptides presented by peptide-MHC complexes on target cells. Therefore, we investigated whether the activity of IgE-specific CTLs was dependent on MHC Class I molecule expression on target cells. IgE-specific peptide IgEp44 is restricted by MHC Class I molecule Db and induces specific CTLs (Fig. 4c). When an anti-Db mAb was added, it completely blocked the recognition of IgEp44/Db complexes and the cytolysis of target cells induced by IgEp44-specific CTLs (Fig. 4c). Hence, the killing activity of IgE-specific CTLs was dependent on the interaction of T cell receptors on CTLs and peptide-MHC complexes expressed on target cells. To further examine the influence of MHC Class I molecule expression on the function of IgE-specific CTLs in vivo, we pre-sensitised WT B6 or β2m-/- mice with OVA.(25) IgE responses were induced in β2m-/- mice as well as WT B6 mice (Fig. 4d). However, adoptively transferred IgE-specific CTLs inhibited IgE responses only in WT mice rather than in β2m-/-mice. Thus, the activity of IgE-specific CTLs is dependent on the expression of MHC Class I molecules, and the recognition of peptide-MHC Class I complexes on target cells is required for the function of IgE-specific CTLs in vivo.
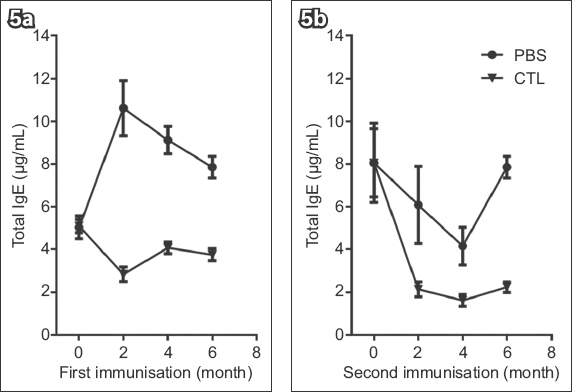
We investigated the long-term in vivo effect of adoptively transferred IgE-specific CTLs in an asthmatic mouse model. Pre-sensitised mice were randomly divided into two groups: one was given IgE-specific CTLs (i.e. CTL group), while the other received PBS (i.e. PBS group) during the initial challenge. At the time intervals indicated, both groups of mice were re-challenged with OVA, and serum IgE was measured one week after each challenge. Before the IgE-specific CTL treatment, IgE levels were elevated in both groups (Fig. 5a). However, as early as two weeks following CTL treatment, IgE responses were markedly inhibited when the mice were re-challenged with OVA (data not shown). The inhibition remained for up to six months in the CTL group (Fig. 5a). In the PBS group, IgE responses recovered four months after receiving CTL treatment (data not shown), and these mice were administered a second IgE-specific CTL treatment (Fig. 5b). Again, IgE responses of both groups were reduced and the resultant low levels of IgE remained up to four months following CTL retreatment (Fig. 5b). In summary, IgE-specific CTLs induced a sustained inhibition of IgE responses in vivo, and those mice with a recovering IgE response could respond to a second CTL treatment with reduced levels of IgE following the antigen challenge.
Fig. 5
Charts show long-term effect of specific cytotoxic T lymphocytes (CTLs) on immunoglobulin E (IgE) response in vivo, including: (a) the primary IgE-specific CTL treatment and (b) the second treatment with IgE-specific CTLs on mice that had recurrent IgE responses. The mice were re-challenged with antigen at the indicated times. IgE responses were measured one week after each challenge. Ten mice received phosphate-buffered saline (PBS) or IgE-specific CTLs.
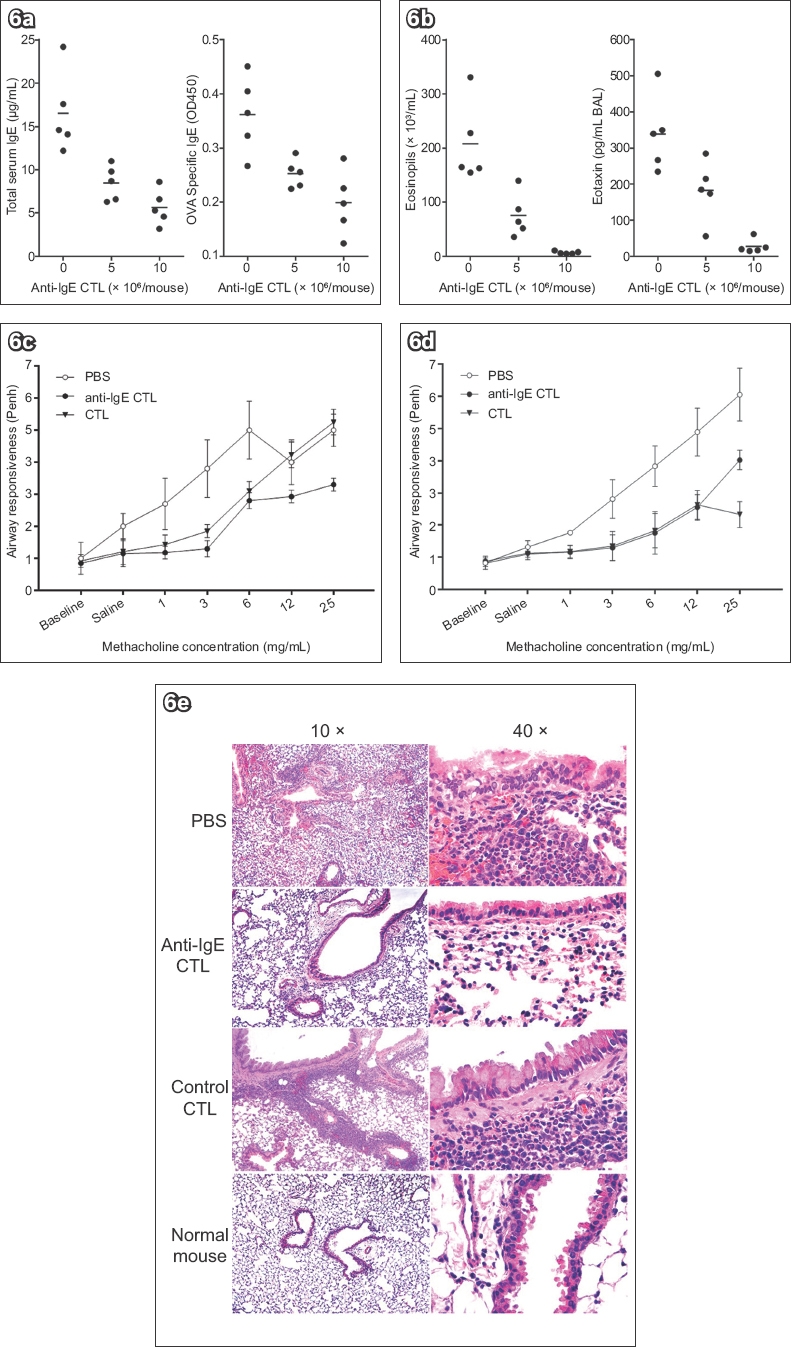
Since IgE-specific CTLs effectively inhibited IgE responses in a mouse model of allergic asthma (Figs. 2–5), we investigated whether airway inflammation could be reduced by IgE-specific CTL treatment. Pre-sensitised mice were treated with indicated doses of IgE-specific CTLs during the intranasal challenges. Two weeks after CTL treatment, both total serum IgE and OVA-specific IgE levels were measured. The IgE level decreased about 50% in the mice that received 5 × 106 IgE-specific CTLs but was inhibited more than 70% in the mice receiving twice that dose of CTLs (Fig. 6a). Five weeks after the CTL treatment, the mice were re-challenged intranasally with OVA every other day, for a total of three times. Mouse BAL fluid was examined 24 hours after the final challenge. Consistent with the inhibition of IgE responses, inflammatory cells in the BAL were dramatically reduced in the CTL-treated mice; this effect was most dramatic for eosinophils (Fig. 6b; partial data not shown). As a major chemokine for eosinophils, eotaxin in the BAL was also measured. The concentration of eotaxin, as well as eosinophils, was significantly reduced in the CTL-treated mice (Fig. 6b). The reduction of eosinophils and eotaxin in the BAL was dependent on the dose of administered CTLs and correlated with the inhibition of IgE responses by specific CTLs (Figs. 6a & b). Thus, IgE-specific CTLs effectively inhibited both IgE responses and airway inflammation in a dose-dependent manner.
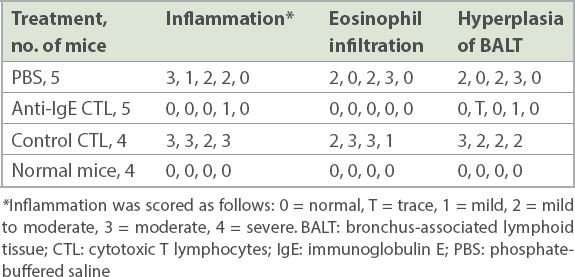
The effect of IgE-specific CTLs on airway inflammation was further confirmed with examination of lung histology (Fig. 6c & Table II). Adult CB6F1/J mice were sensitised and treated with anti-IgE CTL or control CTL during the challenge. The mice were re-challenged five weeks after CTL treatment; BAL and lung tissue were collected from each mouse, and H&E staining was performed. The lung inflammation of each mouse was independently evaluated by a pathologist. Without IgE-specific CTL treatment, sensitised and challenged mice exhibited moderate to severe inflammation with eosinophil infiltration in the lung and hyperplasia of bronchial-associated lymphoid tissue (BALT). However, IgE-specific CTL treatment made all three pathological changes decrease in the lung. Eosinophil infiltration was absent in all of the CTL-treated mice, and mild inflammation and trace to mild hyperplasia of BALT were observed in the lung of some CTL-treated mice. The inhibition of lung inflammation was specific for CTL treatment, since the control CTLs did not inhibit airway inflammation, eosinophil infiltration or hyperplasia of BALT.
Fig. 6
Charts show (a) dose-dependent inhibition of immunoglobulin E (IgE) responses, measured two weeks after treatment with two different doses (5 × 106 and 10 × 106) of IgE-specific cytotoxic T lymphocytes (CTLs); (b) dose-dependent reduction of eosinophils and eotaxin in bronchoalveolar lavage five weeks post IgE-specific CTL treatment; and (c & d) reductions in airway hyperresponsiveness with increasing doses of methacholine, measured using whole body plethysmography, in two independent experiments. (e) Photomicrographs show reductions in airway inflammation following treatment with anti-IgE CTLs (5 × 106/mouse; haematoxylin & eosin). OVA: ovalbumin; PBS: phosphate-buffered saline
Table II
Effect of anti-IgE CTLs on airway inflammation for each mouse.
The hallmarks of asthma are airway hyperresponsiveness (AHR) and airway inflammation.(26) Since IgE-specific CTLs could inhibit IgE responses and reduce airway inflammation, we investigated whether CTL treatment could reduce AHR. Sensitised mice were treated with IgE-specific anti-IgE CTLs and re-challenged as described in Fig. 6b. 24 hours after the last challenge, AHR was assessed with Penh measurements in response to increasing doses of nebulised methacholine using whole body plethysmographs.(27,28) In the absence of IgE-specific CTL treatment, the challenge of pre-sensitised mice induced AHR to an increasing concentration of nebulised methacholine (Figs. 6d & e). However, the induced AHR was markedly reduced in the CTL-treated mice, while the control CTLs had little effect (Fig. 6d). The AHR was significantly reduced by IgE-specific CTLs to a level similar to that in normal mice (Fig. 6e). Thus, IgE-specific CTLs effectively reduced both airway inflammation and AHR in pre-sensitised mice.
DISCUSSION
Immunotherapy has been developed to fight diseases such as cancer, autoimmune diseases and Alzheimer’s disease.(29-31) In this study, we demonstrated that one kind of T cell-based immunotherapy CTLs specific for IgE, a non-tumour self-antigen expressed on a subset of B cells, can be generated in vitro with aAPCs.(16) The in vitro generated IgE-specific CTLs effectively inhibited IgE responses to antigen in pre-sensitised mice, while the IgG responses were almost unaffected. Inhibition of IgE responses by CTLs required expression of both PF by CTLs and MHC Class I molecules on target cells. Enomoto et al has demonstrated that allergen-specific CTLs require PF expression to suppress allergic airway inflammation.(32) In contrast, IFN-γ released by IgE-specific CTLs was not essential for inhibition of the IgE responses. Thus, inhibition of IgE responses by IgE-specific CTLs required both recognition of peptide-MHC Class I complexes and subsequent PF-mediated killing. Apparently, alteration of the balance between Th1 and Th2 responses owing to IFN-γ release from the IgE-specific CTLs did not play a major role in the inhibition of IgE responses in vivo.
Eosinophilic inflammation in the lung is an important feature of allergic asthma.(33) Adoptively transferred IgE-specific CTLs could inhibit both IgE responses and airway eosinophilic inflammation in the animal model of asthma. The inhibition efficiency depended on the dose of IgE-specific CTLs transferred and was directly correlated with the reduction of airway inflammation. This data indicates that specific CTL-mediated inhibition of IgE responses leads to the reduction of airway eosinophilic inflammation.
Anti-IgE antibodies that compete in binding to IgE at the same site as its FcεRI have been developed for the treatment of asthma.(3,4,34) These antibodies inhibit IgE effector function by blocking IgE binding to its high affinity receptors expressed on mast cells and basophils, consequently blocking the inflammatory cascade induced by cross-linking of receptor-bound IgE. One of them, omalizumab (Xolair), has been developed and used successfully for the treatment of patients with moderate to severe asthma.(3,4,11,34,35) Xolair has been demonstrated to improve symptoms and reduce rescue medication and corticosteroid use in patients with allergic asthma.(34,36) However, despite the near-complete reduction of free IgE by the anti-IgE antibody, the total IgE level (IgE-anti-IgE complexes) continued to rise and was shown to be fivefold higher in patients who received anti-IgE for 16 weeks.(3,13) Within weeks after discontinuing the treatment, free IgE level returned to base level,(11) and most of the patients needed treatment with an anti-IgE antibody every 2–4 weeks for an extended period. In our study, IgE-specific CTLs recognised IgE-derived peptides presented by MHC Class I molecules. A single IgE-specific CTL treatment was sufficient to inhibit IgE responses to antigenic challenge for up to six months (Fig. 5), suggesting that a direct inhibition or elimination of IgE-producing B cells and plasma cells by IgE-specific CTLs could be a more efficient approach to the treatment of allergic asthma and some other allergic disorders.
Additionally, IgE-specific CTLs enhanced IgG2a responses to the tested antigens in the sensitised mouse model (Fig. 3c). This enhancement of IgG2a responses appears to be unspecific for IgE-specific CTLs, because the control CTLs that did not inhibit IgE responses could also enhance the IgG2a responses (Fig. 3c). Considering that CTLs produce various cytokines including IFN-g, the enhanced IgG2a responses might reflect a shift towards a Th1 response as a regulation result of cytokines produced by adoptively transferred CTLs. Since regulation of immune responses towards a Th1 response (immune deviation) may be beneficial for allergic disorders, the adoptively transferred IgE-specific CTLs may be helpful for allergic asthma development through two different mechanisms: (a) inhibition of IgE responses by eliminating IgE-producing B cells and plasma cells through recognition of IgE peptide-MHC Class I complexes on target cells, followed by PF-mediated killing; and (b) immune deviation of the allergic response by CTL-produced cytokines.
Production of cytokines by T cells can be regulated by specific co-stimulatory molecules presented on the surface of APC.(37) Co-stimulation of CD8 T cells with APC expressing either CD80 or CD86 molecules induced both IL-2 and IL-4 expression. Interestingly, co-stimulation of CD8 T cells with APC expressing both CD80 and CD54 molecules enhanced gene expression of IL-2 as well as IL-10, whereas IL-4 expression was completely inhibited. Thus, activation of CD8 T cells with APC expressing different co-stimulatory molecules can be used to regulate cytokine production profile. In this study, IgE-specific CTLs were stimulated with APC expressing both CD54 and CD80. As expected, these CTLs express IL-10 rather than IL-4. It has been reported that IL-10 can be used to ameliorate allergen-induced asthma, airway hyper-reactivity and inflammation.(38-41) Thus, regulation of cytokine production by CTLs could be an additional strategy for regulation of immune response in vivo by IgE-specific CTLs.
These novel findings indicate that the central tolerance of T cells to IgE is incomplete and that IgE-specific T cells can be activated in vitro in the presence of optimal stimulation in the mouse model. We have also generated human IgE-specific CTLs ex vivo, and the function of these CTLs has been tested in vitro. Together with studies of CTLs targeting IgE in both mouse and human, these investigations are defining potential clinical applications to enhance CTL function to kill IgE-producing B cells in the airway in asthma cases. Although it is not known if IgE-specific CD8 T cells can be activated in vivo and play a role in the regulation of IgE responses, adoptive transfer of in vitro-generated IgE-specific CTLs can potentially provide a novel tool for the treatment of severe asthma and other IgE-mediated allergic disorders. In addition, generation of CTLs for other self-antigens that are transiently and specifically expressed on cells of the immune system may provide a new approach to immune regulation in vivo.
ACKNOWLEDGEMENTS
This study was funded by Zhejiang Provincial Top Key Discipline of Biology Open Foundation (11610331251507 to Chen J) and Zhejiang Provincial Key Laboratory of Silkworm Bioreactor and Biomedicine, Zhejiang, China.
References Gauvreau GM, Harris JM, Boulet LP, et al. Targeting membrane-expressed IgE B cell receptor with an antibody to the M1 prime epitope reduces IgE production.Sci Transl Med. 2014;6:243-85. Galli SJ, Tsai M.Mast cells in allergy and infection:versatile effector and regulatory cells in innate and adaptive immunity.Eur J Immunol. 2010;40:1843-51. Babu KS, Arshad SH, Holgate ST.Omalizumab, a novel anti-IgE therapy in allergic disorders.Expert Opin Biol Ther. 2001;1:1049-58. Chung KF.Anti-IgE monoclonal antibody, omalizumab:a new treatment for allergic asthma.Expert Opin Pharmacother. 2004;5:439-46. Holgate ST, DjukanoviÄ R, Casale T, Bousquet J.Anti-immunoglobulin E treatment with omalizumab in allergic diseases:an update on anti-inflammatory activity and clinical efficacy.Clin Exp Allergy. 2005;35:408-16. Rafi A, Do LT, Katz R, et al. Effects of omalizumab in patients with food allergy.Allergy Asthma Proc. 2010;31:76-83. Hong CJ, Tsang AC, Quinn JG, et al. Anti-IgE monoclonal antibody therapy for the treatment of chronic rhinosinusitis:a systematic review.Syst Rev. 2015;4:166. Barnes PJ.Cytokine modulators as novel therapies for asthma.Annu Rev Pharmacol Toxicol. 2002;42:81-98. Kaplan AP, Joseph K, Maykut RJ, Geba GP, Zeldin RK.Treatment of chronic autoimmune urticaria with omalizumab.J Allergy Clin Immunol. 2008;122:569-73. Maurer M, Altrichter S, Bieber T, et al. Efficacy and safety of omalizumab in patients with chronic urticaria who exhibit IgE against thyroperoxidase.J Allergy Clin Immunol. 2011;128:202-209e5. Berger WE.Monoclonal anti-IgE antibody:a novel therapy for allergic airways disease.Ann Allergy Asthma Immunol. 2002;88:152-62. 208. Corne J, Djukanovic R, Thomas L, et al. The effect of intravenous administration of a chimeric anti-IgE antibody on serum IgE levels in atopic subjects:efficacy, safety, and pharmacokinetics.J Clin Invest. 1997;99:879-87. Chang TW.The pharmacological basis of anti-IgE therapy.Nat Biotechnol. 2000;18:157-62. Wooldridge L, Clement M, Lissina A, et al. MHC class I molecules with Superenhanced CD8 binding properties bypass the requirement for cognate TCR recognition and nonspecifically activate CTLs.J Immunol. 2010;184:3357-66. Sun S, Cai Z, Langlade-Demoyen P, et al. Dual function of Drosophila cells as APCs for naive CD8+T cells:implications for tumor immunotherapy.Immunity. 1996;4:555-64. Cai Z, Brunmark A, Jackson MR, et al. Transfected Drosophila cells as a probe for defining the minimal requirements for stimulating unprimed CD8+T cells.Proc Natl Acad Sci U S A. 1996;93:14736-41. Cai Z, Kishimoto H, Brunmark A, et al. Requirements for peptide-induced T cell receptor downregulation on naive CD8+T cells.J Exp Med. 1997;185:641-51. Leong KP, Huston DP.Understanding the pathogenesis of allergic asthma using mouse models.Ann Allergy Asthma Immunol. 2001;87:96-110. Parker KC, Bednarek MA, Coligan JE.Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains.J Immunol. 1994;152:163-75. Baas EJ, van Santen HM, Kleijmeer MJ, et al. Peptide-induced stabilization and intracellular localization of empty HLA class I complexes.J Exp Med. 1992;176:147-56. Ortiz-Navarrete V, Hämmerling GJ.Surface appearance and instability of empty H-2 class I molecules under physiological conditions.Proc Natl Acad Sci U S A. 1991;88:3594-7. Kung TT, Jones H, Adams GK, 3rdet al. Characterization of a mane model of allergic pulmonary inflammation.Int Arch Allergy Immunol. 1994;105:83-90. Kägi D, Ledermann B, Bürki K, et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice.Nature. 1994;369:31-7. Dalton DK, Pitts-Meek S, Keshav S, et al. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes.Science. 1993;259:1739-42. Koller BH, Marrack P, Kappler JW, Smithies O.Normal development of mice deficient in beta 2M, MHC class I proteins, and CD8+T cells.Science. 1990;248:1227-30. Wills-Karp M.Immunologic basis of antigen-induced airway hyperresponsiveness.Annu Rev Immunol. 1999;17:255-81. DeLorme MP, Moss OR.Pulmonary function assessment by whole-body plethysmography in restrained versus unrestrained mice.J Pharmacol Toxicol Methods. 2002;47:1-10. Hamelmann E, Schwarze J, Takeda K, et al. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography.Am J Respir Crit Care Med. 1997;156:3 Pt 1766-75. Sabado RL, Bhardwaj N.Cancer immunotherapy:dendritic-cell vaccines on the move.Nature. 2015;519:300-1. Nguyen NT, Raskopf E, Shah-Hosseini K, Zadoyan G, Mösges R.A review of allergoid immunotherapy:is cat allergy a suitable target?.Immunotherapy. 2016;8:331-49. Mulder CK, Dong Y, Brugghe HF, et al. Immunization with small amyloid-b-derived cyclopeptide conjugates diminishes amyloid-b-induced neurodegeneration in mice.J Alzheimers Dis. 2016;52:1111-23. Enomoto N, Hyde E, Ma JZ, et al. Allergen-specific CTL require perforin expression to suppress allergic airway inflammation.J Immunol. 2012;188:1734-41. Possa SS, Leick EA, Prado CM, Martins MA, Tibério IF.Eosinophilic inflammation in allergic asthma.Front Pharmacol. 2013;4:46. Corren J, Casale T, Deniz Y, Ashby M.Omalizumab, a recombinant humanized anti-IgE antibody, reduces asthma-related emergency room visits and hospitalizations in patients with allergic asthma.J Allergy Clin Immunol. 2003;111:87-90. Bhutani M, Yang WH, Hébert J, de Takacsy F, Stril JL.The real world effect of omalizumab add on therapy for patients with moderate to severe allergic asthma:the ASTERIX observational study.PLoS One. 2017;12:e0183869. McNicholl DM, Heaney LG.Omalizumab:the evidence for its place in the treatment of allergic asthma.Core Evid. 2008;3:55-66. Luksch CR, Winqvist O, Ozaki ME, et al. Intercellular adhesion molecule-1 inhibits interleukin 4 production by naive T cells.Proc Natl Acad Sci U S A. 1999;96:3023-8. Akbari O, Dekruyff RH, Umetsu DT.Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen.Nat Immunol. 2001;2:725-31. Oh JW, Seroogy CM, Meyer EH, et al. CD4 T-helper cells engineered to produce IL-10 prevent allergen-induced airway hyperreactivity and inflammation.J Allergy Clin Immunol. 2002;110:460-8. Francis JN, Till SJ, Durham SR.Induction of IL-10+CD4+CD25+T cells by grass pollen immunotherapy.J Allergy Clin Immunol. 2003;111:1255-61. Böhm L, Maxeiner J, Meyer-Martin H, et al. IL-10 and regulatory T cells cooperate in allergen-specific immunotherapy to ameliorate allergic asthma.J Immunol. 2015;194:887-97.
REFERENCES:
1. Gauvreau GM, Harris JM, Boulet LP, et al. Targeting membrane-expressed IgE B cell receptor with an antibody to the M1 prime epitope reduces IgE production. Sci Transl Med 2014; 6:243-85. https://doi.org/10.1126/scitranslmed.3008961
PMid:24990880
2. Galli SJ, Tsai M. Mast cells in allergy and infection: versatile effector and regulatory cells in innate and adaptive immunity. Eur J Immunol 2010; 40:1843-51. https://doi.org/10.1002/eji.201040559
PMid:20583030 PMCid:PMC3581154
3. Babu KS, Arshad SH, Holgate ST. Omalizumab, a novel anti-IgE therapy in allergic disorders. Expert Opin Biol Ther 2001; 1:1049-58. https://doi.org/10.1517/14712598.1.6.1049
PMid:11728235
4. Chung KF. Anti-IgE monoclonal antibody, omalizumab: a new treatment for allergic asthma. Expert Opin Pharmacother 2004; 5:439-46. https://doi.org/10.1517/14656566.5.2.439
PMid:14996639
5. Holgate ST, Djukanović R, Casale T, Bousquet J. Anti-immunoglobulin E treatment with omalizumab in allergic diseases: an update on anti-inflammatory activity and clinical efficacy. Clin Exp Allergy 2005; 35:408-16. https://doi.org/10.1111/j.1365-2222.2005.02191.x
PMid:15836747
6. Rafi A, Do LT, Katz R, et al. Effects of omalizumab in patients with food allergy. Allergy Asthma Proc 2010; 31:76-83. https://doi.org/10.2500/aap.2010.31.3304
PMid:20167148
7. Hong CJ, Tsang AC, Quinn JG, et al. Anti-IgE monoclonal antibody therapy for the treatment of chronic rhinosinusitis: a systematic review. Syst Rev 2015; 4:166. https://doi.org/10.1186/s13643-015-0157-5
PMid:26581392 PMCid:PMC4652338
9. Kaplan AP, Joseph K, Maykut RJ, Geba GP, Zeldin RK. Treatment of chronic autoimmune urticaria with omalizumab. J Allergy Clin Immunol 2008; 122:569-73. 10. Maurer M, Altrichter S, Bieber T, et al. Efficacy and safety of omalizumab in patients with chronic urticaria who exhibit IgE against thyroperoxidase. J Allergy Clin Immunol 2011; 128:202-209.e5.
11. Berger WE. Monoclonal anti-IgE antibody: a novel therapy for allergic airways disease. Ann Allergy Asthma Immunol 2002; 88:152-62, 208. https://doi.org/10.1016/S1081-1206(10)61989-8
12. Corne J, Djukanovic R, Thomas L, et al. The effect of intravenous administration of a chimeric anti-IgE antibody on serum IgE levels in atopic subjects: efficacy, safety, and pharmacokinetics. J Clin Invest 1997; 99:879-87. https://doi.org/10.1172/JCI119252
PMid:9062345 PMCid:PMC507895
13. Chang TW. The pharmacological basis of anti-IgE therapy. Nat Biotechnol 2000; 18:157-62. https://doi.org/10.1038/72601
PMid:10657120
14. Wooldridge L, Clement M, Lissina A, et al. MHC class I molecules with Superenhanced CD8 binding properties bypass the requirement for cognate TCR recognition and nonspecifically activate CTLs. J Immunol 2010; 184:3357-66. 15. Sun S, Cai Z, Langlade-Demoyen P, et al. Dual function of Drosophila cells as APCs for naive CD8+ T cells: implications for tumor immunotherapy. Immunity 1996; 4:555-64. https://doi.org/10.1016/S1074-7613(00)80482-3
16. Cai Z, Brunmark A, Jackson MR, et al. Transfected Drosophila cells as a probe for defining the minimal requirements for stimulating unprimed CD8+ T cells. Proc Natl Acad Sci U S A 1996; 93:14736-41. https://doi.org/10.1073/pnas.93.25.14736
PMid:8962124 PMCid:PMC26205
17. Cai Z, Kishimoto H, Brunmark A, et al. Requirements for peptide-induced T cell receptor downregulation on naive CD8+ T cells. J Exp Med 1997; 185:641-51. 18. Leong KP, Huston DP. Understanding the pathogenesis of allergic asthma using mouse models. Ann Allergy Asthma Immunol 2001; 87:96-110. https://doi.org/10.1016/S1081-1206(10)62201-6
19. Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol 1994; 152:163-75.
20. Baas EJ, van Santen HM, Kleijmeer MJ, et al. Peptide-induced stabilization and intracellular localization of empty HLA class I complexes. J Exp Med 1992; 176:147-56. https://doi.org/10.1084/jem.176.1.147
PMid:1613456 PMCid:PMC2119279
21. Ortiz-Navarrete V, Hämmerling GJ. Surface appearance and instability of empty H-2 class I molecules under physiological conditions. Proc Natl Acad Sci U S A 1991; 88:3594-7. https://doi.org/10.1073/pnas.88.9.3594
PMid:2023907 PMCid:PMC51498
22. Kung TT, Jones H, Adams GK 3rd, et al. Characterization of a murine model of allergic pulmonary inflammation. Int Arch Allergy Immunol 1994; 105:83-90. https://doi.org/10.1159/000236807
PMid:8086833
23. Kägi D, Ledermann B, Bürki K, et al. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 1994; 369:31-7. https://doi.org/10.1038/369031a0
PMid:8164737
24. Dalton DK, Pitts-Meek S, Keshav S, et al. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science 1993; 259:1739-42. https://doi.org/10.1126/science.8456300
PMid:8456300
25. Koller BH, Marrack P, Kappler JW, Smithies O. Normal development of mice deficient in beta 2M, MHC class I proteins, and CD8+ T cells. Science 1990; 248:1227-30. https://doi.org/10.1126/science.2112266
PMid:2112266
27. DeLorme MP, Moss OR. Pulmonary function assessment by whole-body plethysmography in restrained versus unrestrained mice. J Pharmacol Toxicol Methods 2002; 47:1-10. https://doi.org/10.1016/S1056-8719(02)00191-0
28. Hamelmann E, Schwarze J, Takeda K, et al. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med 1997; 156(3 Pt 1):766-75. https://doi.org/10.1164/ajrccm.156.3.9606031
PMid:9309991
29. Sabado RL, Bhardwaj N. Cancer immunotherapy: dendritic-cell vaccines on the move. Nature 2015; 519:300-1. https://doi.org/10.1038/nature14211
PMid:25762139
30. Nguyen NT, Raskopf E, Shah-Hosseini K, Zadoyan G, Mösges R. A review of allergoid immunotherapy: is cat allergy a suitable target? Immunotherapy 2016; 8:331-49. https://doi.org/10.2217/imt.15.121
PMid:26860435
31. Mulder CK, Dong Y, Brugghe HF, et al. Immunization with small amyloid-b-derived cyclopeptide conjugates diminishes amyloid-b-induced neurodegeneration in mice. J Alzheimers Dis 2016; 52:1111-23. https://doi.org/10.3233/JAD-151136
PMid:27060957 PMCid:PMC4927839
32. Enomoto N, Hyde E, Ma JZ, et al. Allergen-specific CTL require perforin expression to suppress allergic airway inflammation. J Immunol 2012; 188:1734-41. https://doi.org/10.4049/jimmunol.1102699
PMid:22250087
33. Possa SS, Leick EA, Prado CM, Martins MA, Tibério IF. Eosinophilic inflammation in allergic asthma. Front Pharmacol 2013; 4:46. https://doi.org/10.3389/fphar.2013.00046
PMid:23616768 PMCid:PMC3627984
34. Corren J, Casale T, Deniz Y, Ashby M. Omalizumab, a recombinant humanized anti-IgE antibody, reduces asthma-related emergency room visits and hospitalizations in patients with allergic asthma. J Allergy Clin Immunol 2003; 111:87-90. https://doi.org/10.1067/mai.2003.49
PMid:12532101
35. Bhutani M, Yang WH, Hébert J, de Takacsy F, Stril JL. The real world effect of omalizumab add on therapy for patients with moderate to severe allergic asthma: the ASTERIX observational study. PLoS One 2017; 12:e0183869. https://doi.org/10.1371/journal.pone.0183869
PMid:28859150 PMCid:PMC5578673
36. McNicholl DM, Heaney LG. Omalizumab: the evidence for its place in the treatment of allergic asthma. Core Evid 2008; 3:55-66.
37. Luksch CR, Winqvist O, Ozaki ME, et al. Intercellular adhesion molecule-1 inhibits interleukin 4 production by naive T cells. Proc Natl Acad Sci U S A 1999; 96:3023-8. https://doi.org/10.1073/pnas.96.6.3023
PMid:10077630 PMCid:PMC15888
38. Akbari O, Dekruyff RH, Umetsu DT. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat Immunol 2001; 2:725-31. https://doi.org/10.1038/90667
PMid:11477409
39. Oh JW, Seroogy CM, Meyer EH, et al. CD4 T-helper cells engineered to produce IL-10 prevent allergen-induced airway hyperreactivity and inflammation. J Allergy Clin Immunol 2002; 110:460-8. https://doi.org/10.1067/mai.2002.127512
PMid:12209095
40. Francis JN, Till SJ, Durham SR. Induction of IL-10+CD4+CD25+ T cells by grass pollen immunotherapy. J Allergy Clin Immunol 2003; 111:1255-61. https://doi.org/10.1067/mai.2003.1570
PMid:12789226
41. Böhm L, Maxeiner J, Meyer-Martin H, et al. IL-10 and regulatory T cells cooperate in allergen-specific immunotherapy to ameliorate allergic asthma. J Immunol 2015; 194:887-97. https://doi.org/10.4049/jimmunol.1401612
PMid:25527785