

Abstract
INTRODUCTION
This study aimed to assess the kind of haematological abnormalities that are present in patients with mitochondrial disorders (MIDs) and the frequency of their occurrence.
METHODS
The blood cell counts of a cohort of patients with syndromic and non-syndromic MIDs were retrospectively reviewed. MIDs were classified as ‘definite’, ‘probable’ or ‘possible’ according to clinical presentation, instrumental findings, immunohistological findings on muscle biopsy, biochemical abnormalities of the respiratory chain and/or the results of genetic studies. Patients who had medical conditions other than MID that account for the haematological abnormalities were excluded.
RESULTS
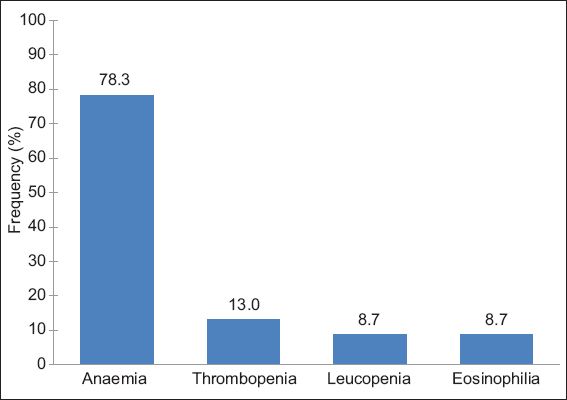
A total of 46 patients (‘definite’ = 5; ‘probable’ = 9; ‘possible’ = 32) had haematological abnormalities attributable to MIDs. The most frequent haematological abnormality in patients with MIDs was anaemia. 27 patients had anaemia as their sole haematological problem. Anaemia was associated with thrombopenia (n = 4), thrombocytosis (n = 2), leucopenia (n = 2), and eosinophilia (n = 1). Anaemia was hypochromic and normocytic in 27 patients, hypochromic and microcytic in six patients, hyperchromic and macrocytic in two patients, and normochromic and microcytic in one patient. Among the 46 patients with a mitochondrial haematological abnormality, 78.3% had anaemia, 13.0% had thrombopenia, 8.7% had leucopenia and 8.7% had eosinophilia, alone or in combination with other haematological abnormalities.
CONCLUSION
MID should be considered if a patient’s abnormal blood cell counts (particularly those associated with anaemia, thrombopenia, leucopenia or eosinophilia) cannot be explained by established causes. Abnormal blood cell counts may be the sole manifestation of MID or a collateral feature of a multisystem problem.
INTRODUCTION
Mitochondrial disorders (MIDs) are an increasingly recognised group of mono- or multisystem metabolic disorders that manifest clinically in either syndromic or non-syndromic forms.(1) MIDs are caused by mutations in either mitochondrial DNA (mtDNA)- or nuclear DNA (nDNA)-located genes encoding subunits of respiratory chain complexes, ancillary proteins of respiratory chain complexes, transfer RNA (tRNA), ribosomal RNA (rRNA), proteins involved in the mtDNA replication machinery, the coenzyme-Q pathway, the mitochondrial transport machinery or the mitochondrial biosynthesis.(2) MIDs frequently manifest in the central nervous system, peripheral nervous system, endocrine system, heart, retina, cochlea, liver and/or kidneys (mitochondrial multiorgan disorder syndrome [MIMODS]).(3) MIDs rarely manifest in the gastrointestinal tract, cartilage, integument or the bone marrow (resulting in haematological abnormalities).(4) They can be either syndromic (e.g. mitochondrial encephalomyopathy lactic acidosis and stroke-like episodes [MELAS]; Kearns-Sayre syndrome; neuropathy, ataxia and retinitis pigmentosa; Pearson syndrome)(2) or non-syndromic. Haematological abnormalities may occur together with syndromic or non-syndromic MIDs.
The present study had the following aims: (a) identify the haematological abnormalities that are present in a large cohort of patients with MIDs; (b) determine the frequency with which haematological abnormalities occur in patients with MIDs; and (c) collate information on the treatments that were applied to these patients.
METHODS
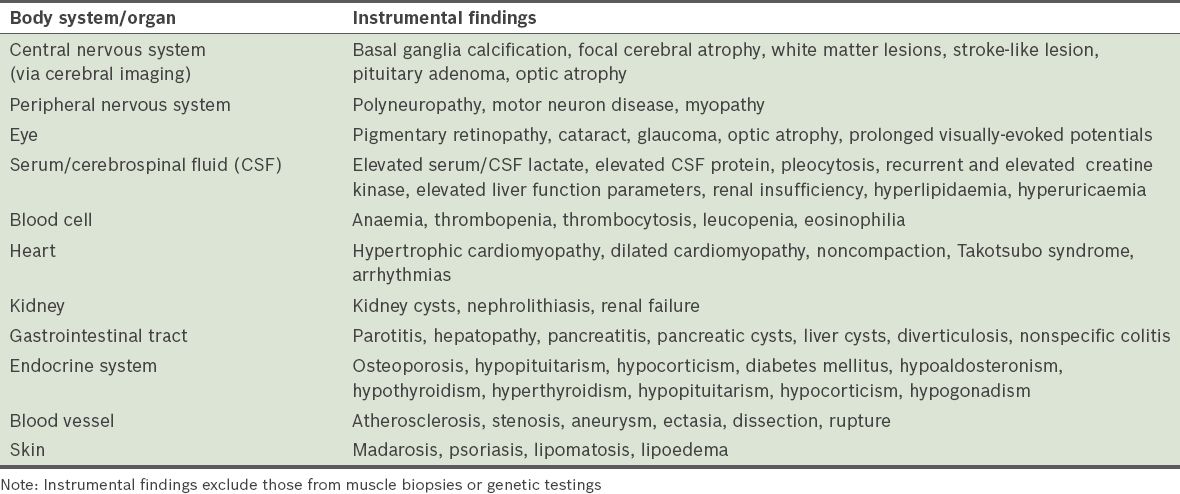
We retrospectively reviewed the blood cell counts of patients with syndromic or non-syndromic MIDs who attended the inpatient or outpatient units of the Krankenanstalt Rudolfstiftung, Austria, between May 2008 and April 2012. MID was classified as: (a) ‘definite’ if the clinical presentation was indicative of MID and there was biochemical or genetic evidence of a mitochondrial defect; (b) ‘probable’ if the clinical presentation was indicative of MID and muscle biopsy findings showed immunohistological abnormalities (e.g. cytochrome-c oxidase [COX]-negative fibres, ragged-red fibres or succinate dehydrogenase hyper-reactive fibres), or if electron microscopy showed abnormally shaped or structured mitochondria with or without paracristalline inclusions, glycogen or fat depositions; and (c) ‘possible’ if the clinical presentation suggested MID and instrumental findings (other than muscle biopsy) were indicative of MID. This categorisation was in accordance with guidelines of the European Federation of Neurological Sciences.(2) Clinical findings and instrumental findings (other than those from muscle biopsy) that were suggestive of MID are summarised in Tables
Table I
Symptoms and signs suggestive of a mitochondrial disorder.

Table II
Instrumental findings indicative of a mitochondrial disorder.
Only patients who had repeated abnormal blood cell counts that could not be attributed to causes other than MID were included in the analysis. Exclusion of patients with haematological abnormalities attributed to other causes was carried out by an experienced haematologist. Patients who had anaemia due to inadequate erythrocyte production, increased erythrocyte destruction, acute or chronic bleeding, and/or fluid overload (
Table III
Causes of anaemia that were excluded before it was attributed to a mitochondrial disorder.
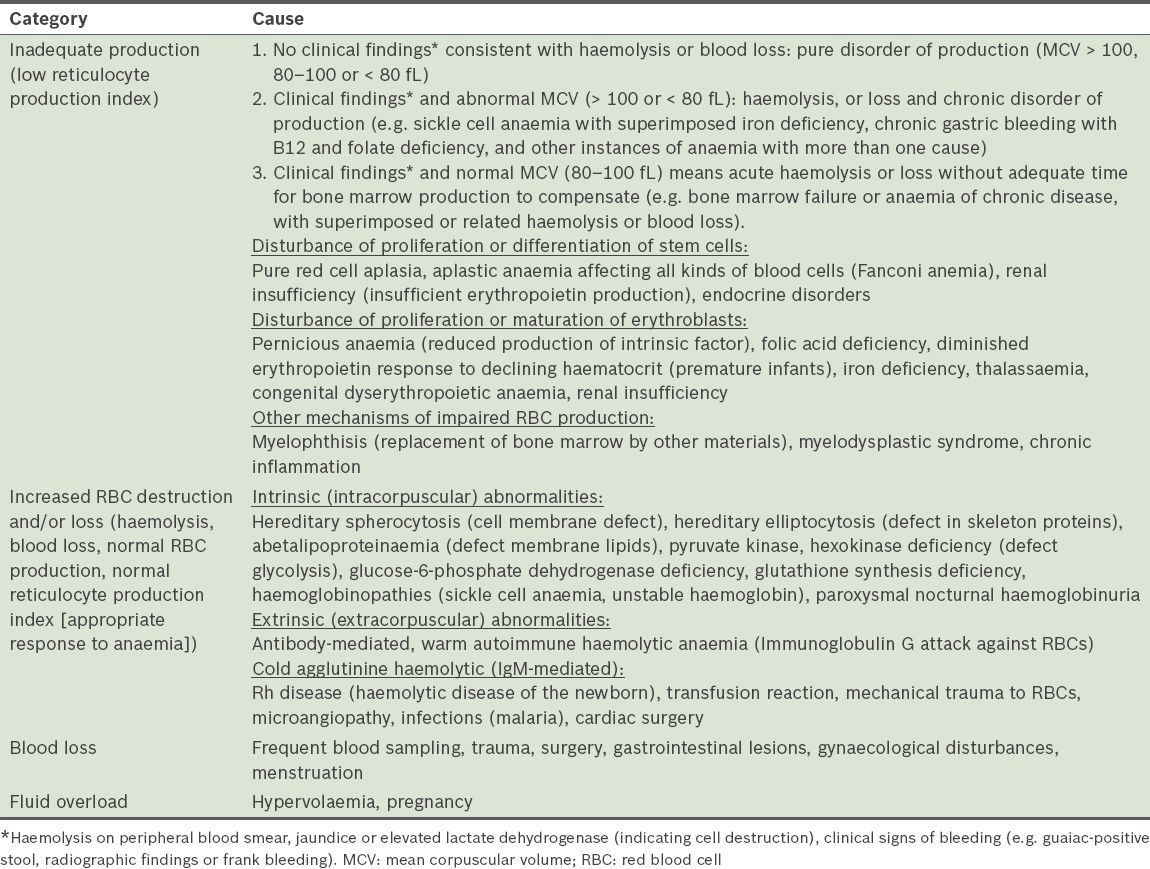
Table IV
Causes of thrombopenia that were excluded before it was attributed to a mitochondrial disorder.

Table V
Causes of thrombocytosis that were excluded before it was attributed to a mitochondrial disorder.

Table VI
Causes of leucopenia that were excluded before it was attributed to a mitochondrial disorder.

RESULTS
During the four-year observational period, 444 patients were diagnosed with MID – 15 (3.4%) ‘definite’, 54 (12.2%) ‘probable’ and 375 (84.5%) ‘possible’ MIDs. Stratification of the patients and the results of their haematological investigations are summarised in
Table VII
Stratification and results of haematological investigations of patients with mitochondrial disorders (MIDs).
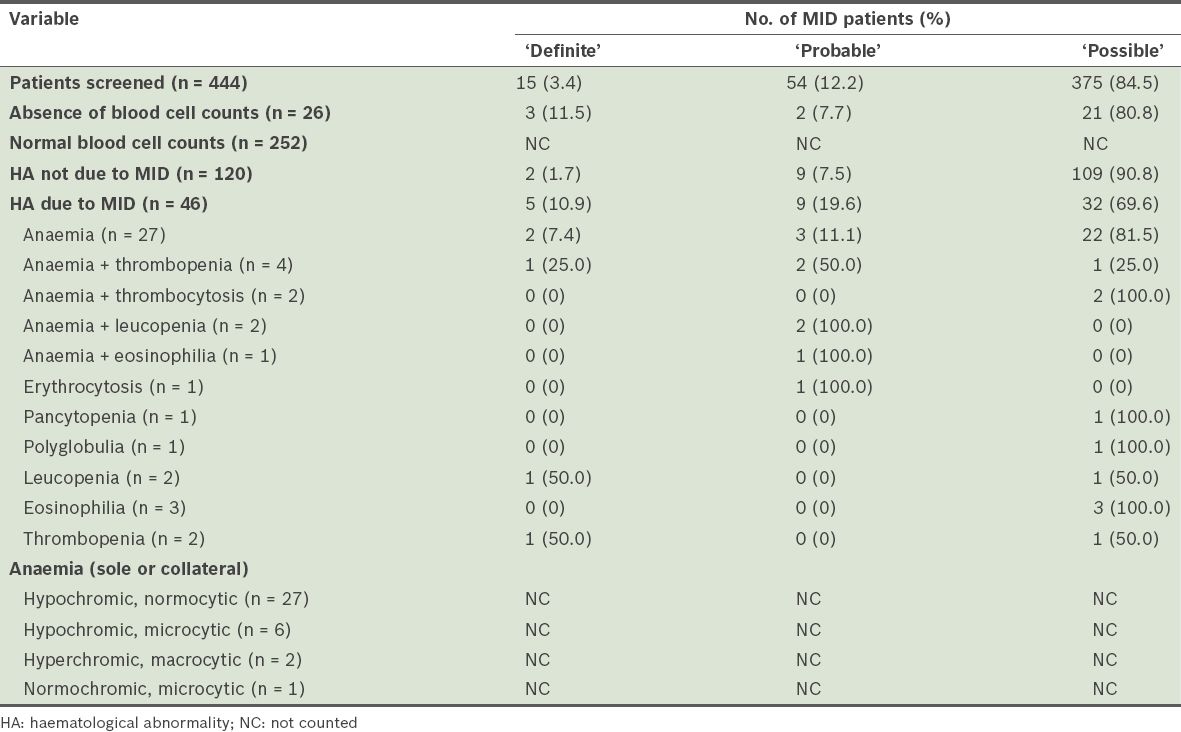
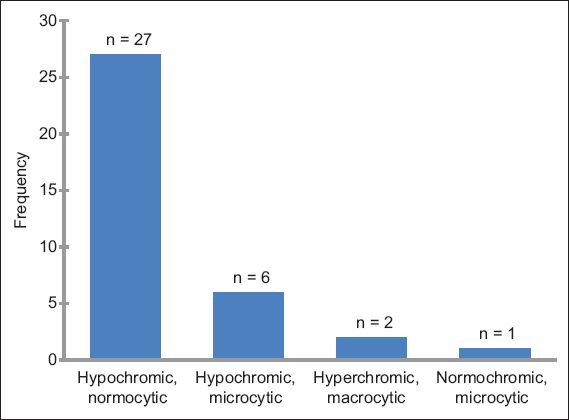
Of the 46 patients with MID-related haematological abnormalities, 27 (2 ‘definite’, 3 ‘probable’ and 22 ‘possible’ MIDs) had anaemia only. Anaemia was associated with thrombopenia in four patients, thrombocytosis in two patients, leucopenia in two patients, and eosinophilia in one patient. Other sole haematological abnormalities attributable to MID included: erythrocytosis (n = 1); pancytopenia (n = 1); polyglobulia (n = 1); thrombopenia (n = 2); leucopenia (n = 2); and eosinophilia (n = 3). None of the patients had thrombocytosis as the sole haematological problem. Alone, or in combination, anaemia was present in 36 patients, thrombopenia in six patients, leucopenia in four patients, erythrocytosis in one patient, thrombocytosis in two patients and eosinophilia in four patients. Among patients with anaemia, the reticulocyte count was available for three patients and elevated in one patient (data not shown). Anaemia was hypochromic and normocytic in 27 patients, hypochromic and microcytic in six patients, hyperchromic and macrocytic in two patients, and normochromic and microcytic in one patient (
Fig. 1
Bar graph shows the frequency of anaemia types among the 46 patients with haematological abnormalities attributable to mitochondrial disorder.
Anaemia was the most frequent haematological abnormality among patients who had haematological abnormalities attributable to MID. Anaemia attributable to MID was found in 8.6% of the 418 patients who had available blood cell counts, while thrombopenia and leucopenia attributable to MID was found in 1.4% and 1.0% of the 418 patients, respectively. Among the 46 patients who had haematological abnormalities attributable to MID, 78.3% had anaemia, 13.0% had thrombopenia, 8.7% had leucopenia and 8.7% had eosinophilia, alone or in combination with other haematological abnormalities (
Fig. 2
Bar graph shows the frequency of anaemia, thrombopenia, leucopenia and eosinophilia among the 46 patients with haematological abnormalities attributable to mitochondrial disorder.
Except for anaemia, the haematological abnormalities found in the 46 patients did not require medical treatment. Anaemia was resistant to treatment with folic acid, pyridoxine, vitamin B12 and iron in all 36 patients, while one patient required blood transfusions. Erythropoietin was attempted in one patient, but it did not yield long-term success. None of the patients with thrombopenia required drug treatment or infusion with thrombocyte concentrates. A patient with thrombocytosis received hydroxyurea from time to time.
DISCUSSION
The present study shows that haematological abnormalities in syndromic and non-syndromic MIDs frequently include anaemia, thrombopenia, leucopenia and eosinophilia, and less frequently include thrombocytosis, erythrocytosis, polyglobulia, and pancytopenia, in combination or alone. The most common of these abnormalities was anaemia, followed by thrombopenia and leucopenia. Treatment for anaemia, which included vitamin and iron substitution, blood transfusions and/or erythropoietin, was applied only in some patients.
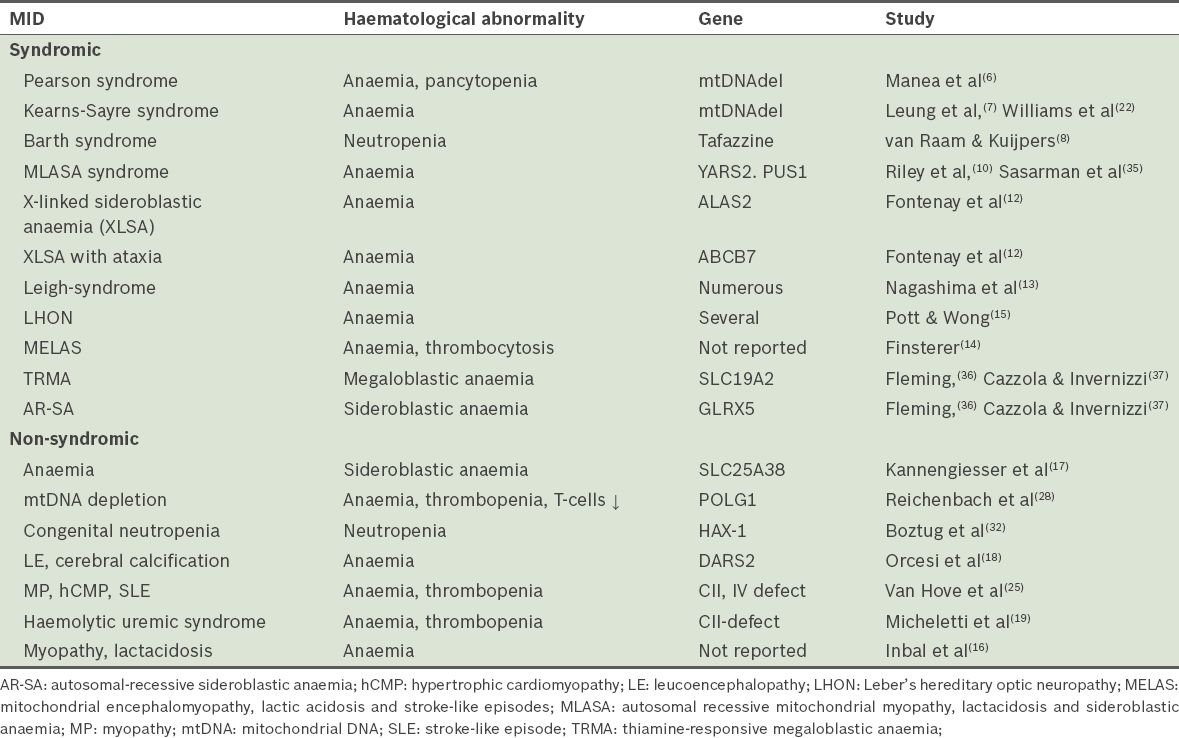
Haematological abnormalities are well-known dominant or collateral features of MIDs.(4) Syndromic MIDs with haematological manifestations include: (a) Pearson syndrome, a rare congenital disorder with severe sideroblastic anaemia or pancytopenia and exocrine pancreas insufficiency;(6) occasionally, the kidneys, liver, guts or skin are affected; (b) Kearns-Sayre syndrome, a disorder characterised by onset at < 20 years, ophthalmoplegia, pigmentary retinopathy, conduction block, cerebellar ataxia, increased cerebrospinal fluid protein and/or sideroblastic anaemia;(7) (c) Barth syndrome, a disorder characterised by noncompaction, mitochondrial myopathy and neutropenia;(8) (d) autosomal recessive mitochondrial myopathy, lactic acidosis and sideroblastic anaemia (MLASA);(9,10) (e) X-linked sideroblastic anaemia (XLSA), the most common type of sideroblastic anaemia;(11,12) and (f) X-linked sideroblastic anaemia with ataxia (XLSA-A)(12) (
Table VIII
Mitochondrial disorders associated with haematological abnormalities.
Haematological abnormalities occur in MIDs because of the involvement of mitochondria in haematopoiesis (through oxidative phosphorylation, heme synthesis, iron metabolism, iron-sulfur [Fe-S] cluster biogenesis and apoptosis).(12) Mutations in the ALAS2 gene, which encodes the 5-aminolevulinate-synthase-2 (ALAS-2) isoform that catalyses the first step in heme synthesis in the mitochondrial matrix, lead to ineffective erythropoiesis, a characteristic feature of XLSA. Mutations in the adenosine triphosphate-binding cassette protein B7 (ABCB7), which is identified in XLSA-A, disrupt the maturation of cytosolic Fe-S clusters, leading to mitochondrial iron accumulation.(12) Although it is unknown why tafazzin mutations (which lead to Barth-syndrome) are associated with leucopenia, the involvement of tafazzin in the cardiolipin metabolism process may be a possible cause.(8) Mutations in the YARS-2 gene, which codes for mitochondrial tyrosyl-tRNA synthetase, causes reduced aminoacylation, resulting in decreased protein synthesis, respiratory chain dysfunction and MLASA.(10) Large-scale deletions in the mtDNA, whose integrity depends on a specific DNA polymerase, are hallmarks of Pearson and Kearns-Sayre syndromes. In the early stages of acquired myelodysplastic syndromes, exacerbation of physiological pathways involving caspases and mitochondria in erythroid differentiation leads to abnormal activation of a mitochondrion-mediated apoptotic cell death pathway.(12) mtDNA deletions may induce abnormalities in erythropoiesis by causing haematopoietic cell-specific respiration defects.(20) The high frequency of haematological abnormalities in patients who had definite MID in our study may indicate that haematological abnormalities are indeed a phenotypic feature of MIDs.
In the present study, anaemia was the most frequent haematological abnormality in syndromic and non-syndromic MIDs. This finding is consistent with other reports, where anaemia was also more frequent in MIDs than abnormalities in thrombocyte or leucocyte count (either as the sole, dominant or collateral feature of the phenotype) (
Sideroblastic anaemia is heterogeneous and characterised by excess iron accumulation in the mitochondria of erythroblasts.(11,26) The morphological hallmark of sideroblastic anaemia is the ring sideroblast, a pathological erythroid precursor containing an excessive amount of non-heme iron deposits in the mitochondria with perinuclear distribution, generating a ring appearance.(26) Sideroblastic anaemia may be hereditary or acquired. Hereditary sideroblastic anaemia may be caused by mutations in nDNA-located genes such as ALAS2, ABCB7, GRLX5, SLC25A38, SF3B1, JAK2, TET2, MPLW515 and YARS2, or by large mtDNA deletions.(26) Acquired sideroblastic anaemia can be primary (refractory anaemia with ring sideroblasts [RARS]), representing a subtype of myelodysplastic syndrome, or secondary (due to drugs, toxins, copper deficiency or malignancy). Ring sideroblasts may develop as a result of defective heme synthesis in erythroblasts (as in XLSA), a defect in the Fe-S cluster assembly, release of Fe-S protein precursor from the mitochondria (as in XLSA-A) or defective intracellular iron metabolism in erythroid cells (as in RARS).(11,26)
Thrombopenia has been occasionally reported in patients with MIDs. Although thrombopenia is more frequently seen in Pearson syndrome,(6) it can also occur in Kearns-Sayre syndrome.(27) In non-syndromic MIDs, thrombopenia has been reported in a boy who had a mtDNA depletion syndrome due to a POLG1 mutation; he also presented with immunodeficiency with severe recurrent infections, anaemia, automutilation and cerebral abnormalities.(28) Thrombopenia was also a phenotypic feature in a 40-year-old woman with myopathy, renal insufficiency, cardiomyopathy, stroke-like episodes and anaemia.(25) The present study provides evidence that thrombopenia is a less frequent phenotypic feature of MIDs than expected. Thrombopenia particularly occurs in patients with non-syndromic MIDs. Since some patients with MIDs develop hypothyroidism (a common cause of thrombopenia),(29) thyroid disorders and other possible causes (
Leucopenia as the sole haematological abnormality of MID has been reported in syndromic and non-syndromic MIDs.(30) Leucopenia in the form of neutropenia is commonly seen in patients with Barth syndrome, where neutropenia is the hallmark of the phenotype.(31) Neutropenia does not necessarily result in an increased rate of infections. Leucopenia (including neutropenia), together with thrombopenia, may also be a feature of Kearns-Sayre syndrome.(27) One of the possible causes of severe congenital neutropenia in non-syndromic MID is mutation in the gene encoding the mitochondrial protein HAX-1, a regulator of calcium signalling and apoptosis progression.(32) Neutropenia in non-syndromic MID may also be caused by mtDNA depletion due to a POLG1 mutation, which may manifest as fatal, neonatal-onset T-cell immunodeficiency that is associated with chronic infection.(28) Leucopenia as a feature of pancytopenia has been reported in patients with Pearson and Kearns-Sayre syndromes.(27,33)
Eosinophilia is a rare phenotypic feature of MIDs that has rarely been reported in the literature. Finsterer and Höger reported eosinophilia in a 65-year-old woman (who had pituitary adenoma, basal ganglia calcification, tetraspasticity, restless legs, hypoacusis, tinnitus, atrial fibrillation, myopathy, polyneuropathy, polyarthralgia, osteoporosis, hyponatraemia and hyperlipidaemia), which was attributed to non-syndromic, multisystem MID after a muscle biopsy and exclusion of Churg-Strauss syndrome.(34) She also presented with a history of recurrent thrombopenia in addition to eosinophilia. MID was diagnosed based on the patient’s clinical presentation and muscle biopsy findings (single ragged-red fibres, COX-negative fibres and slight increase in glycogen).(34) However, in the present study, eosinophilia attributable to MID was not infrequent; it was found in 11 patients, four of whom also had other haematological abnormalities.
Therapeutic options depend on the type of blood cell abnormality. Options include substitution of iron, pyridoxine (SLC25A38), blood transfusions, thrombocyte transfusions, administration of erythropoietin, hydroxylurea, leucocyte stimulation factor and haematopoetic stem cell transplantation (i.e. bone marrow transplantation) in patients with Pearson syndrome.(17,24) In the present study, the application of anaemia treatment to the patients was largely ineffective.
To conclude, MID should be considered a cause of abnormal blood cell count, especially if the abnormality cannot be explained by established causes or there is unexplained anaemia, thrombopenia, thrombocytosis, leucopenia or eosinophilia (alone or in combination). Abnormal blood cell counts may be the sole manifestation of an MID but, more frequently, it is part of a multisystem problem. Haematological abnormalities may be the initial manifestation of an MID.