

Abstract
INTRODUCTION
The current gold standard for diagnosing interstitial lung disease (ILD) involves an ILD clinic evaluation, followed by discussion in a multidisciplinary meeting (MDM). However, there is a paucity of data on the impact of ILD MDMs on the diagnosis and management of ILDs in Southeast Asia. We studied the clinical impact of the ILD service on the diagnosis and management of ILDs at a university-affiliated tertiary hospital in Singapore.
METHODS
A single-centre retrospective review was done on 97 consecutive patients referred for evaluation to the ILD service from March 2016 to August 2017.
RESULTS
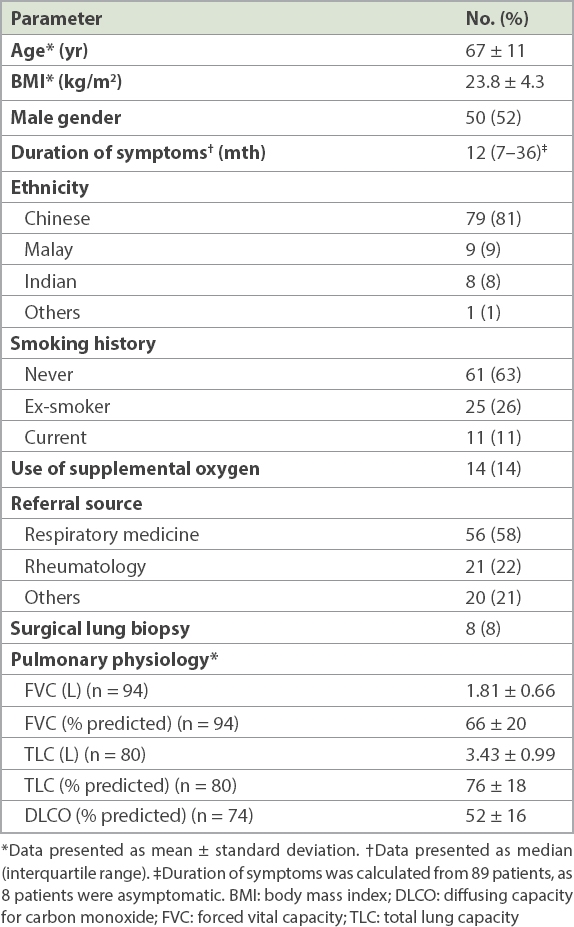
Mean age of the patients was 67 ± 11 years. Gender distribution was almost equal (52% male), with a majority of never-smokers (63%). Mean forced vital capacity (FVC) was 1.81 ± 0.66 L (66% ± 20% predicted). The three commonest referral diagnoses were ILD of uncertain classification (n = 38, 39%), connective tissue disease-associated ILD (CTD-ILD) (n = 24, 25%) and idiopathic pulmonary fibrosis (IPF) (n = 16, 17%). Following evaluation by the ILD service, there was a change of diagnosis in 60 (62%) patients and a change of management in 71 (73%) patients. The majority of consensus MDM diagnoses were IPF (n = 35, 36%), CTD-ILD (n = 30, 30%) and others (n = 15, 15%). There was a significant prognostic separation between the IPF and non-IPF diagnoses made following evaluation by the ILD service.
CONCLUSION
The ILD service allowed for more precise subtyping of various ILDs. This is particularly useful for IPF patients, who can benefit from antifibrotic therapies.
INTRODUCTION
Interstitial lung disease (ILD) is a heterogeneous group of diseases with varying disease behaviours and prognoses. Multidisciplinary meetings (MDMs) that involve chest clinicians, radiologists and pathologists are the current diagnostic standard for ILD.(1) Flaherty et al first showed that multidisciplinary discussions involving the different subspecialties improve diagnostic performance in patients with ILDs.(2) Two societies have recently published guidance on how an MDM should be conducted,(3,4) with an ontological framework being developed to standardise the diagnostic classification of the different ILDs.(5)
Diagnosing idiopathic pulmonary fibrosis (IPF), the most common form of idiopathic interstitial pneumonia (IIP), has become critically important, as the disease has a devastating prognosis with a median survival of 2.5–3.5 years.(6) There are also management implications, as patients with IPF can now potentially be treated with new antifibrotic therapies such as pirfenidone and nintedanib, both of which were approved in Singapore in 2016 following two landmark trials published in 2014(7,8) that showed a reduction in the decline of lung function in IPF patients. In addition, immunosuppression with a combination of prednisolone and azathioprine should be avoided, as it increases mortality and hospitalisation in this group of patients.(9) In contrast, patients with certain types of connective tissue disease-associated interstitial lung disease (CTD-ILD), such as systemic sclerosis and idiopathic inflammatory myopathies-related ILDs, may benefit from systemic immunomodulatory therapies like cyclophosphamide and mycophenolate mofetil.
Studies on ILD MDM that showed positive diagnostic and management implications on patient care are predominantly from established ILD expert centres in the United States,(2) Europe(10,11) and Australia.(12) There is a paucity of such data from Southeast Asia. We performed a retrospective evaluation of our centre’s ILD service to evaluate whether the positive impact of an ILD clinic and MDM on the diagnosis and management of ILDs demonstrated in overseas expert centres can be translated to our local setting. In addition, we validated the diagnostic accuracy of IPF in our ILD service against other non-IPF diagnoses.
METHODS
We are a university-affiliated tertiary hospital in Singapore with an ILD multidisciplinary team that comprises a chest physician, two thoracic radiologists, a rheumatologist and a pathologist, all of whom have received prior training from overseas expert ILD centres. In addition, we have a clinical pharmacist who provides disease and medication counselling. We performed a single-centre retrospective review of consecutive patients referred to the ILD clinic with subsequent discussions in ILD MDMs over an 18-month period from March 2016 to August 2017. All patients underwent evaluation with a complete history, physical examination and standardised investigations. The investigations included basic blood tests, lung function tests, high-resolution computed tomography of the chest, autoimmune serologies and serum brain natriuretic peptide levels. In selected patients, echocardiography, bronchoalveolar lavage, transbronchial lung biopsy or surgical lung biopsy (SLB) were performed.
Cases of suspected IPF, non-IPF fibrotic lung disease (such as fibrotic nonspecific interstitial pneumonia [NSIP], chronic hypersensitivity pneumonitis [HP] and sarcoidosis), ILDs that were not fully characterised and those with complex management issues were presented at our monthly ILD MDM. During the MDM, all the available clinical information was projected on a screen and the relevant thoracic radiology images were presented by the radiologists. The pathologist also presented selected biopsy images during the meeting. The referral diagnosis and management plan as stated in the referral letter were then compared against the consensus diagnosis that was made at the MDM. Diagnoses were classified according to the American Thoracic Society/European Respiratory Society IIP guidelines.(1)
We validated the diagnoses made in the ILD MDM by comparing the mortality of IPF against the other diagnoses. The survival period was calculated from the referral date to the ILD clinic to the date of death or 1 December 2017, which was the end of the study period. This study (no. 2017/00001) was reviewed by the National Healthcare Group Domain Specific Review Board (DSRB) and the need for DSRB review was deemed not necessary.
We used IBM SPSS Statistics version 20.0 for Windows (IBM Corp, Armonk, NY, USA) for all statistical analyses. Categorical data was displayed as absolute numbers and relative frequencies. Missing data was removed from the denominator when calculating relative frequencies. Continuous data was shown as mean ± standard deviation for normally distributed data or as median (interquartile range [IQR]) for nonparametric data. Survival analysis was used to validate the MDM diagnosis. The Kaplan-Meier survival curve was utilised to show cumulative survival of IPF versus non-IPF MDM diagnoses. Log-rank test for survival equality was used to compare survival curves. Cox regression analysis was used to evaluate the hazard ratio (95% confidence interval [CI]) of MDM IPF diagnoses. Statistical significance was determined at p < 0.05.
RESULTS
A total of 149 patients were referred to the ILD clinic over an 18-month period from 1 March 2016 to 31 August 2017 (
Fig. 1
Flowchart shows patients referred to the interstitial lung disease (ILD) clinic and subsequent multidisciplinary meeting (MDM) evaluation.
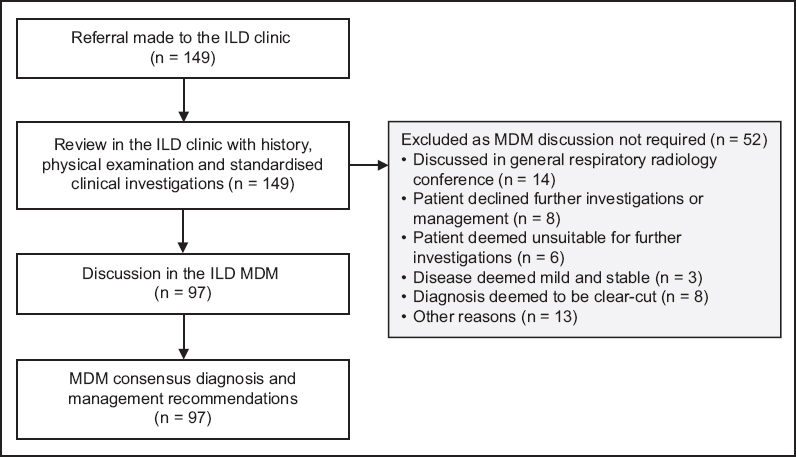
Table I
Baseline demographics and pulmonary physiology (n = 97).
The referral diagnoses and MDM consensus diagnoses are shown in
Fig. 2
Pie charts show (a) referral and (b) multidisciplinary consensus diagnoses. CTD-ILD: connective tissue disease-associated interstitial lung disease; HP: hypersensitivity pneumonitis; ILD: interstitial lung disease; IPAF: interstitial pneumonia with autoimmune features; IPF: idiopathic pulmonary fibrosis; NSIP: nonspecific interstitial pneumonia
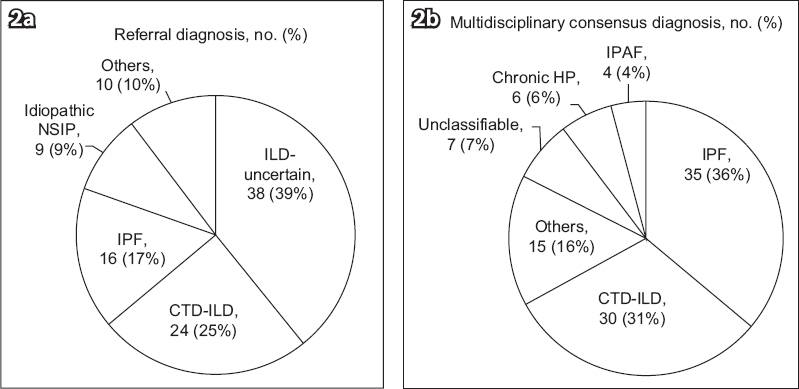
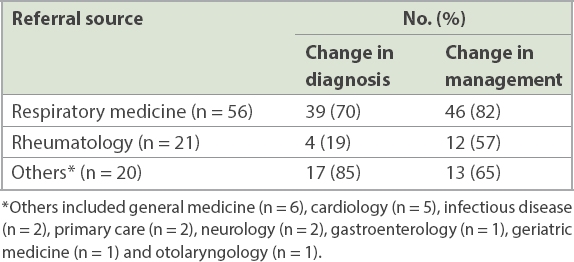
Table II
Change in diagnosis and management according to referral source.
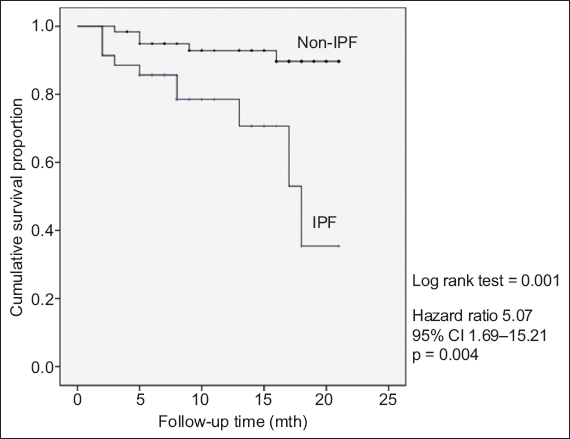
The MDM diagnoses of IPF versus the non-IPF diagnoses were validated in the univariate Cox regression analysis, and the Kaplan-Meier survival curve is shown in
Fig. 3
Kaplan-Meier curve shows survival difference between patients with idiopathic pulmonary fibrosis (IPF) and those without IPF. Median follow-up time was 11 (interquartile range 7–16.5) months. CI: confidence interval
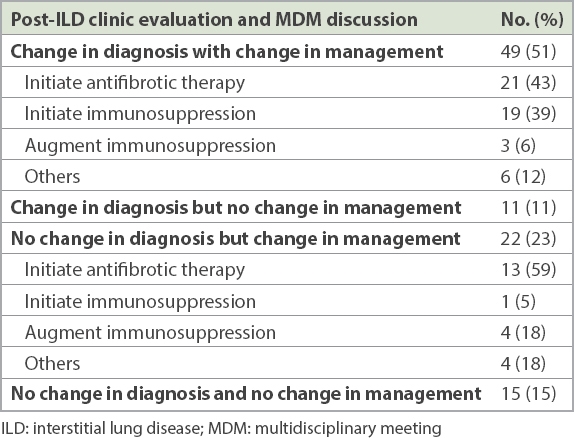
Overall, 71 (73%) patients had a change in management after evaluation by the ILD clinic and MDM. The majority (49/97, 51%) were directly due to a change in diagnosis (
Table III
Changes in management after ILD clinic evaluation and MDM discussion.
DISCUSSION
In this retrospective single-tertiary-centre analysis, a dedicated ILD service comprising an ILD clinic and MDM resulted in changes in diagnosis and subsequent management in the majority of the cohort. In addition, the accuracy of IPF diagnosis was verified by the significant prognostic separation of an IPF diagnosis versus the non-IPF ones. This compares favourably to the studies from Australia and the United Kingdom (UK) in which 53%(12) and 76%(11) of the patients, respectively, had a change in diagnosis after evaluation by an ILD multidisciplinary service. In the UK study, 40% of the patients also had their management altered after ILD multidisciplinary evaluation.(11)
In the present study, the patients who presented to our ILD service had more severe lung disease compared to other similar cohorts. Using FVC as an indicator of disease severity, the mean FVC in our study was 66% predicted, which is lower than the Australian (FVC 77% predicted)(12) and Danish (FVC 71% predicted) cohorts.(13) Also, 14% of our patients were on supplemental oxygen, indicating underlying chronic hypoxaemic respiratory failure or significant exercise desaturation. We speculate that patients presented to our ILD service later in their disease course due to delayed recognition and referral to the appropriate specialists. Indeed, the median duration of symptoms experienced by patients before attendance at the ILD service was one year; this is similar to a European survey of IPF patients where 58% of the respondents reported a delay of more than one year between initial presentation and definite diagnosis of IPF.(14) Hence, further improvement in educating physicians to recognise ILDs is needed so that appropriate referrals can be made promptly.
The main referral diagnosis we received was ILD of an uncertain classification. This mirrors the retrospective analysis of an ILD multidisciplinary service done in the UK(11) where 23% of the ILDs referred were not classified. In our study, the majority of these patients had an eventual diagnosis of IPF. It must be emphasised that the diagnosis of IPF carries major prognostic and management implications. Similarly, patients with underlying CTD-ILD may experience the pulmonary and extra-pulmonary benefits of immunomodulatory treatment, which can control disease activity and prevent progressive organ damage. With ready access to a service that comprises an ILD clinic and MDM, it will no longer be acceptable to label patients with an umbrella term of ILD without further classification, thereby subjecting them to diagnostic and prognostic uncertainties. This may ultimately lead to inappropriate or missed treatment opportunities.(15)
Our SLB rates were much lower than that of the Danish cohort (40%)(13) but higher than that of the UK cohort (5%).(11) This may reflect a change in practice over the years, with more widespread adoption of ILD MDMs so that clinical data, radiological patterns, bronchoalveolar lavage, disease course and treatment outcomes(16) are integrated and discussed among the various specialties. This is reflected in the falling rates of SLB for the diagnosis of IPF; in earlier studies, SLB rates as high as 65% were reported in the 2004 GIPF-001 study(17) versus the more recent 2014 INPULSIS study, where only 22% of the patients underwent SLB.(8) Close to half of our patients were unable to undergo SLB either because they were too old or frail, or the procedure was deemed too high risk. Indeed, SLB carries a 30-day mortality of 2.4% based on a hospital statistics database from 1997 to 2008 in England, UK.(18) This is comparable to the 2.3% 30-day mortality for lobectomy for non-small cell lung cancer,(19) a potentially curative procedure rather than a diagnostic one. Looking forward, transbronchial lung cryobiopsy, which offers an overall diagnostic yield of 81% with lower complication and mortality rates than SLB,(20) may be considered in patients who are unfit for SLB.
Regarding the diagnosis of IPF, only 11 of the 16 patients referred for IPF had an eventual consensus diagnosis of IPF. The ILD clinic and MDM also diagnosed an additional 24 patients with IPF. Even though a recent study, whose participants comprised expert ILD clinicians, suggested that clinicians can diagnose IPF with similar levels of accuracy as their respective MDMs,(10) our results clearly showed that this cannot be replicated in a broader group of chest physicians, each with different levels of experience. Indeed, a subsequent international study evaluating the diagnostic accuracy of a clinical diagnosis of IPF without a multidisciplinary evaluation found that only clinicians with more than 20 years of experience and those with regular MDM attendance were able to diagnose IPF with similar accuracy as IPF experts.(21)
With regard to the distribution of MDM diagnosis, IPF was the most common diagnosis. This is consistent with the findings of the Australian(12) and Danish(13) cohorts. The second most common diagnosis was CTD-ILD, accounting for 30% of the diagnoses. The proportion of this diagnosis is higher compared to the Australian study,(12) the Indian ILD registry(22) and the Denmark ILD cohort,(13) because our centre has a large affiliated rheumatology department.(23) On the contrary, chronic HP and unclassifiable ILD were less frequent in our cohort as compared to other cohorts. The diagnosis of chronic HP is often challenging and 50% or more of patients may not have a clear exposure history.(24) Unlike IPF, there is a lack of consensus guidelines on how this disease is diagnosed and the diagnostic agreement among expert multidisciplinary teams is poor (weighted kappa value of 0.29).(10) With the absence of local data, it remains uncertain if the low frequency represents a true low incidence of this entity in our country or underdiagnosis. The frequency of unclassifiable ILD in our cohort was only 7%, which is lower than the reported frequency of 10%–14%;(13,25) this may be because our referred cases had less complexity than other cohorts that evaluated patients in designated specialised ILD centres.(12,13)
Idiopathic NSIP is a very rare condition, and none of our patients had this diagnosis. Indeed, following rigorous multidisciplinary evaluation by expert clinicians, radiologists and pathologists of an American Thoracic Society Workgroup, only 67 definite or probable idiopathic NSIP patients were identified among 305 patients who were previously reported to have idiopathic NSIP; the majority of the NSIP cases excluded by this workgroup were HP, usual interstitial pneumonia and organising pneumonia.(26) Additionally, NSIP may be a manifestation of an underlying CTD.(27) In our study, patients referred for idiopathic NSIP were reassigned a diagnosis of CTD-ILD, chronic HP, IPF or unclassifiable ILD. This is similar to the existing published literature.
The use of the survival difference between IPF and non-IPF to validate the diagnostic accuracy of IPF at our MDM warrants further discussion. IPF carries a very poor prognosis, which differentiates this disease from other ILDs. In fact, its five-year survival is worse than the majority of malignancies except lung and pancreatic cancers.(28) Median survival is 2.5–3.5 years,(6) and only 21% of the patients demonstrate a slowly progressive course.(29) The survival is markedly worse compared to other ILDs such as chronic HP (median survival seven years)(30) and idiopathic NSIP (five-year survival 82.3%).(26) Similarly, in systemic sclerosis-related ILD, one of the most commonly seen CTD-ILDs, the median survival is 5–8 years.(31) This method has been used to evaluate the diagnostic accuracy of IPF in an international ILD MDM study.(10) The significant prognostic separation between the diagnosis of IPF and other ILDs validates our MDM evaluation in terms of the diagnostic accuracy of IPF, which is the commonest IIP.
Almost three-quarters of our patients had a change in management after the ILD clinic evaluation; this was directly related to more accurate diagnoses in the majority of patients. Management recommendations included initiating antifibrotic or immunomodulatory therapy. There were also a significant number of patients with a change in management without any change in diagnosis, with the predominant treatment recommendation being initiation of antifibrotic therapy. This may be due to general chest physicians’ lack of familiarity with the use of recently approved antifibrotic therapies, as well as the prohibitive costs of these therapies locally. A similar finding is shown in a survey of Canadian academic and community chest physicians or trainees, where only 34% of the responders used the antifibrotic pirfenidone in patients with IPF.(32)
The majority of changes in diagnosis occurred in referrals from respiratory medicine and other disciplines. While it is not surprising to see a high frequency of diagnosis changes among patients referred from other non-respiratory disciplines, it is surprising that almost three-quarters of the referrals from respiratory medicine had a change in diagnosis following evaluation by the ILD service. This may be because the clinicians were unfamiliar with what to look out for in the diagnosis of ILD. Just looking at IPF alone, the commonest of the IIPs, one survey of French pulmonologists found that one-third of the respondents were not aware of the 2011 IPF international guidelines.(33) In another case cohort study, individual academic physicians needed more than 20 years of experience in order to diagnose IPF at a level achieved by expert ILD clinicians.(21) In our study, among the referrals from rheumatologists, a diagnosis of CTD-ILD was much less of an issue; however, after ILD service evaluation, there was a change of management in 57% of the referrals. Although this is lower than the results reported by Castelino et al,(34) their findings must be interpreted in the context of a much higher rate of lung biopsy (40%), which resulted in a change in diagnosis or treatment in 50% of them. It is uncertain if we can achieve similar rates of treatment change if our cohort were to have such a high lung biopsy rate.
There are a few limitations in our study. Firstly, this is a single-centre retrospective study with a small sample size, and the diagnostic accuracy of the MDM can only be validated by the prognostic difference of IPF compared to the non-IPF entities. The logical next step is for an expert centre to validate the consensus MDM diagnosis, a method that was employed by the ILD registry in India.(22) Secondly, the magnitude of the change in diagnosis and management is likely to be overestimated, as the majority of the referral diagnoses were ILD of an uncertain classification. Nonetheless, the ILD service has achieved its aim in providing a more precise diagnosis and management for this group of patients, who would have otherwise been labelled with an uncertain ILD diagnosis. Thirdly, the length of follow-up is relatively short and, consequently, the clinical impact of the change in diagnosis and management cannot be fully characterised. Fourthly, there may be referral bias, as patients who were deemed unfit for antifibrotic therapy or immunomodulation may not have been referred to the ILD service; also, a minority of patients from the ILD clinic were discussed at the general respiratory radiology meeting and not the ILD MDM. Finally, our SLB rates were lower than those reported in the published literature,(13) and so it is uncertain if a greater magnitude of change in diagnosis or management can be achieved with higher biopsy rates.
In conclusion, this is the first study in Southeast Asia to evaluate the clinical impact of a dedicated ILD clinic and MDM. A significant number of patients had a change in diagnosis with subsequent change in management. Importantly, the group with ILD of uncertain classification were subtyped more accurately, with the majority eventually classified as IPF. Patients usually present late in their disease course, so it is important that physicians identify these patients early, so that appropriate referral can be made and specific therapies instituted promptly. Although long-term outcomes remain to be seen, a multidisciplinary approach to diagnosis and management of ILDs should be the standard of care for this group of patients.
ACKNOWLEDGEMENTS
The authors thank Dr Tan Geak Poh and his staff at the respiratory function laboratory, Tan Tock Seng Hospital, Singapore, for their invaluable input to the manuscript and for providing the pulmonary function results. All authors report no conflict of interests.