

Abstract
INTRODUCTION
Childhood immune thrombocytopenia (ITP) remains a diagnosis of exclusion when isolated thrombocytopenia is not part of another disease process. In practice, the diagnosis of ITP can only be confirmed when thrombocytopenia resolves or is excluded after the recognition of a primary cause.
METHODS
The records of 87 consecutive children with isolated thrombocytopenia seen over a nine-year period in a private paediatric haematology practice were reviewed retrospectively. Children in whom a primary cause was eventually found were the subjects of a further descriptive study.
RESULTS
9 (10%) children with isolated thrombocytopenia were not diagnosed with ITP because a primary disease was found. Of these nine cases, four had thrombocytopenia recognised during the neonatal period, consisting of perinatal cytomegalovirus infection (n = 2), meconium aspiration pneumonia (n = 1) and transient abnormal myelopoiesis associated with Down syndrome (n = 1). The remaining five children were each found to have familial thrombocytopenia, portal hypertension, cutaneous mastocytosis, May-Hegglin anomaly and systemic lupus erythematosus. Two of them had a history of failure of response to corticosteroid therapy.
CONCLUSION
Secondary thrombocytopenia is not uncommon in a tertiary paediatric specialty practice with adequate evaluation. Thrombocytopenia occurring during the newborn period and failure of steroid therapy are predictive of secondary cases.
INTRODUCTION
Immune thrombocytopenia (ITP) is a relatively common childhood illness, with an annual incidence of 1.9–6.4 per 100,000 children.(1) A diagnosis of ITP can be made when isolated thrombocytopenia (platelet count < 100 × 109/L) occurs in the absence of identifiable and specific precipitants.(2) All current clinical practice guidelines have recommended a minimum evaluative process to look for secondary causes of thrombocytopenia before a diagnosis of ITP is made.(2,3) Medical literature has numerous examples of ITP mimics of infectious,(4) immune,(5) haematologic,(6) endocrine,(7) neoplastic(8) and other disorders.(9) In practice, however, the frequency of secondary thrombocytopenia has not been systematically reported. Indeed, five major series reporting a total of 3,736 (50–1,784 per series) paediatric patients with thrombocytopenia from America, Asia and Europe in the last eight years did not mention secondary cases.(10-14) A chart review was therefore carried out to see how often secondary thrombocytopenia presents with an ITP-like illness.
METHODS
This retrospective chart review studied children from birth to 18 years of age who were attending a private paediatric haematology practice in Singapore from 2007 to 2015. The practice received local patients as well as patients from overseas, on an inpatient or outpatient basis. The clinic database registered all children as having isolated thrombocytopenia (platelet count < 100 × 109/L) when they presented with bleeding symptoms associated with thrombocytopenia or when they were referred for evaluation of thrombocytopenia with no definitive diagnosis. Children with mild anaemia (haemoglobin [Hb] > 9.0 g/dL) due to recent blood loss, iron deficiency or the thalassaemia trait, as well as children with leucocytosis concurrent with a febrile illness, were included. Cases were excluded if the referring physicians or the reporting laboratory had suspected an alternative diagnosis, such as leukaemia that was later confirmed, or if there was anaemia with Hb ≤ 9.0 g/dL or leucopenia.
All patients were evaluated with a thorough history taking, including familial predispositions, physical examination with particular attention to cutaneous eruptions and organ enlargements, and full blood counts with examination of the peripheral blood smear by a haematologist. The microscopic examination followed a checklist for isolated thrombocytopenia: red cell changes (schistocytes, Howell-Jolly bodies and malarial parasites), leucocyte changes (blasts, atypical lymphocytes, Döhle bodies and Pelger-Huët anomaly) and platelet changes (small platelets, giant platelets, platelet clumps and satellitism). Secondary thrombocytopenia was diagnosed when the isolated thrombocytopenia was determined to be part of another disease process. Children with secondary thrombocytopenia formed the subjects of a descriptive study.
RESULTS
During the study period, 87 consecutive children presented with isolated thrombocytopenia. Among them, five patients presented with thrombocytopenia during the first month of life. There were 32 patients from Singapore and 55 patients of other nationalities. 78 children were diagnosed with ITP. 41 (47.1%) of the 87 children had acute ITP with resolution of thrombocytopenia within three months of diagnosis, 9 (10.3%) had persistent ITP with disease resolution 3–12 months after diagnosis, and 14 (16.1%) had chronic ITP with thrombocytopenia lasting over 12 months. The other 14 (16.1%) children had otherwise typical clinical and laboratory features of ITP but did not have adequate follow-up information for classification.
Secondary thrombocytopenia was diagnosed in 9 (10.3%) children – 4 (80.0%) from the five neonatal onset cases and 5 (6.1%) among the 82 older children. According to nationality, secondary thrombocytopenia was diagnosed in 4 (12.5%) local patients and in 5 (9.1%) foreign patients. Their clinical features are summarised in
Table I
Summary of the cases of secondary thrombocytopenia.
Two neonates were diagnosed with congenital/perinatal cytomegalovirus infection based on the presence of viral genome in the bloodstream. A newborn with Down syndrome was found to have hepatosplenomegaly and presence of megakaryoblasts on the blood film and was thus diagnosed with transient abnormal myelopoiesis. Another neonate was diagnosed with meconium aspiration pneumonia. Thrombocytopenia eventually resolved in these babies.
Among the older children with secondary thrombocytopenia, there were two familial cases. A seven-year-old child with mild thrombocytopenia had a strong family history indicative of an autosomal dominant form of familial thrombocytopenia. A two-year-old girl with mild thrombocytopenia failed to respond to prednisolone therapy prescribed by her primary physician and was found to have May-Hegglin anomaly on the blood smear. Family screening identified the same macrothrombocytopenia and presence of Döhle bodies in her asymptomatic father.
Secondary thrombocytopenia was also found in a two-year-old girl who had a history of recurrent urticarial and pigmented skin eruptions consistent with cutaneous mastocytosis. She had relapsing thrombocytopenia during infections with almost normal counts in between the episodes. A three-year-old presented with a history of persistent thrombocytopenia of six months’ duration and an isolated episode of anaemia following an episode of haematemesis. Prednisolone treatment was discontinued, as there was no response. He was well when he visited for a secondary opinion, and a small palpable spleen was the only positive sign on examination. Portal hypertension was suspected and was confirmed when oesophageal varices were found on upper endoscopy. The remaining patient was an eight-year-old girl who had frequent relapsing thrombocytopenia following successful treatment with prednisolone. A history of nonspecific facial rashes led to positive findings for antinuclear antibodies and anti-DNA antibodies.
DISCUSSION
In an unselected series of isolated thrombocytopenia in a specialist clinic, ITP remains the most frequent diagnosis in the great majority of cases after thorough evaluation. However, secondary thrombocytopenia is not uncommon and accounts for 10% of the cases in this series. Hence, clinicians following up with children who have isolated thrombocytopenia should be alert for alternative diagnoses other than ITP until the disease remits (
Box 1
Causes of childhood thrombocytopenia that mimic immune thrombocytopenia.

The importance of enquiring for a familial history of bleeding or thrombocytopenic disorder cannot be overemphasised. A history of chronic ITP in a first-degree relative may be an important clue, as misdiagnosis of familial thrombocytopenia is common. Parental screening may be indicated at times. As clinical practice guidelines(2,3) and an abundance of case reports(4-9) have highlighted, when ordering additional tests and diagnostic procedures to look for other causes of thrombocytopenia, it is important to take note of atypical events from the patient’s history and abnormal signs other than bruises on physical examination. In this respect, neonatal onset thrombocytopenia represents a distinct clinical entity, and failure to respond to prednisolone therapy can be another warning sign of non-immune causes of isolated thrombocytopenia.
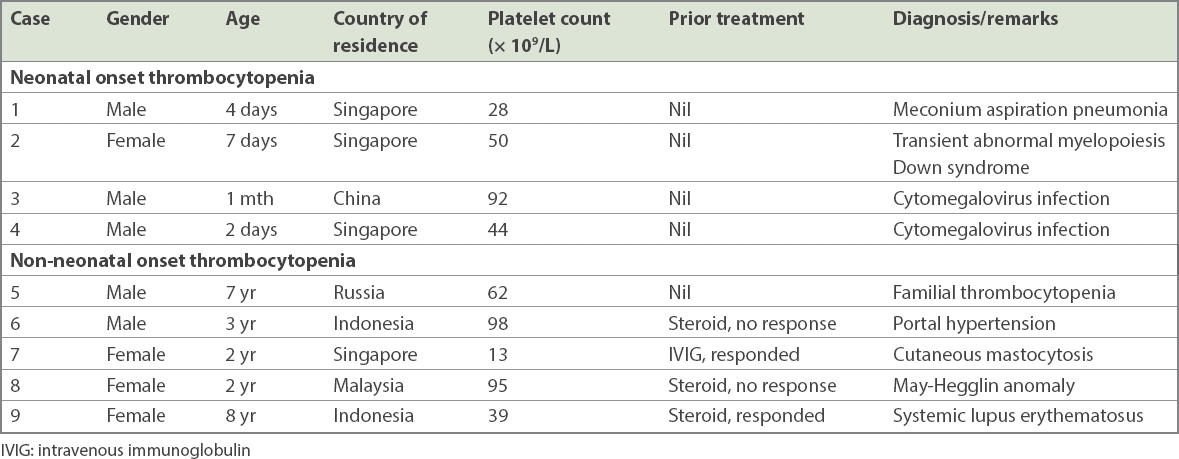
Examination of the peripheral blood smear is an important procedure in the evaluation of isolated thrombocytopenia. The two cases of perinatal cytomegalovirus infection were identified because of atypical lymphocytosis found on the blood film (
Fig. 1
Photomicrograph of the peripheral blood film (Giemsa stain × 100) of a patient with perinatal cytomegalovirus infection shows an atypical lymphocyte (AL) and a normal, small lymphocyte (SL).
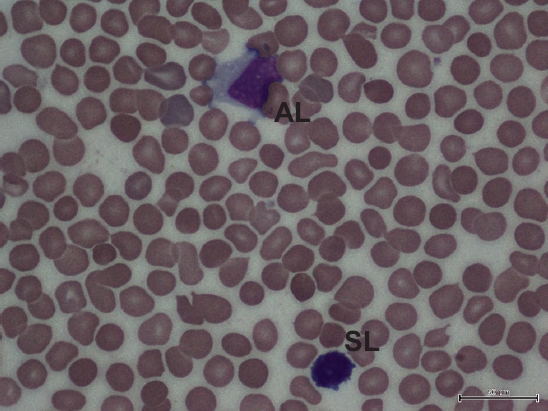
Fig. 2
Photomicrograph of the peripheral blood film (Giemsa stain × 100) of a patient with neonatal transient abnormal myelopoiesis shows two large-sized blasts. The blast on the left side has a blebby cytoplasmic border suggestive of a megakaryoblastic origin.
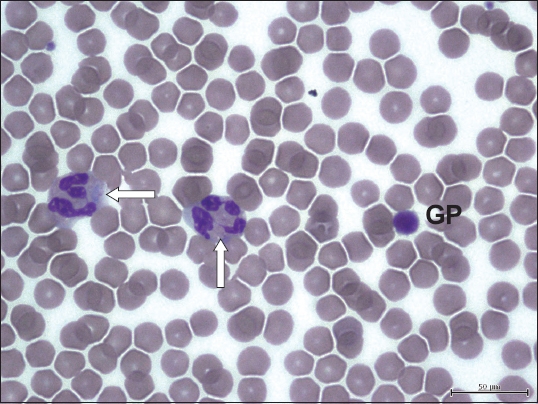
Fig. 3
Photomicrograph of the peripheral blood film (Giemsa stain × 100) of a patient with May-Hegglin anomaly shows two neutrophils with cytoplasmic basophilic inclusions (Döhle bodies, arrows) alongside a giant platelet (GP) that is approximately the size of a red cell.
The main limitation of this study was patient referral bias. As a private practice, it receives patients from a more affluent background, which may have restricted the full spectrum of thrombocytopenic illnesses. On the other hand, a mix of international patients may have introduced clinical entities associated with thrombocytopenia that have not been previously noted in the indigenous population.(17) As alluded to earlier, it is unclear if the patients diagnosed with chronic ITP or those who are lost to follow-up truly have immune thrombocytopenia, because the disease does not have a specific laboratory marker.
In conclusion, secondary thrombocytopenia is not an uncommon diagnosis among children presenting with isolated thrombocytopenia and should be looked out for at the initial diagnosis and during the subsequent follow-up until thrombocytopenia resolves.