

Abstract
INTRODUCTION
Endocrine dysfunction due to iron overload secondary to frequent blood transfusions is a common complication in children with transfusion-dependent thalassaemia (TDT). We ascertained the prevalence of endocrine dysfunction in children with TDT seen in a hospital setting in Malaysia.
METHODS
We reviewed all patients with TDT who had ≥ 8 blood transfusions per year. Patients who had a history of stem cell transplantation, concurrent autoimmune diseases or were newly diagnosed to have TDT were excluded. Standard diagnostic criteria were used in the diagnosis of various endocrine dysfunctions.
RESULTS
Of the 82 patients with TDT, 65% had at least one endocrine dysfunction. Short stature was the commonest (40.2%), followed by pubertal disorders (14.6%), hypoparathyroidism (12.3%), vitamin D deficiency (10.1%), hypocortisolism (7.3%), diabetes mellitus (5.2%) and overt hypothyroidism (4.9%). Subclinical hypothyroidism and pre-diabetes mellitus were seen in 13.4% and 8.6% of the patients, respectively. For children aged < 10 years, the prevalence of both thyroid dysfunction and hypoparathyroidism was 9.1%.
CONCLUSION
Two-thirds of children with TDT experienced at least one endocrine dysfunction. Thyroid dysfunction and hypoparathyroidism may be missed if endocrine screening is only performed in children with TDT > 10 years of age. Close monitoring for endocrine dysfunction and hormonal therapy is essential to prevent long-term adverse outcomes.
INTRODUCTION
Frequent blood transfusions in patients with transfusion-dependent thalassaemia (TDT) may lead to various morbidities and mortality due to excessive iron deposition. To date, improved survival is seen in patients with TDT when the target level of serum ferritin is below 2,500 ng/mL.(1) Because lifespans have increased, more and more TDT patients with endocrine dysfunctions secondary to chronic iron overload have been diagnosed.(2) Nearly all endocrine organs, including the anterior pituitary gland, thyroid, pancreas and parathyroid glands, are at risk of haemochromatosis. The two most prevalent endocrine dysfunctions encountered are short stature and pubertal disorders.(3)
It is estimated that approximately 150–350 infants with thalassaemia are born in Malaysia each year.(4) With the availability of safe blood transfusion and iron chelation therapy, the survival of patients with TDT in Malaysia has improved significantly. However, haematopoietic stem cell transplantation (HSCT), which is curative for TDT, is not widely available in the country. Thus, medical management of patients with TDT, including endocrine dysfunctions, is a pressing medical challenge. To the best of our knowledge, the present study is the first to ascertain the prevalence and clinical presentation of endocrine dysfunctions in a cohort of children with TDT managed in a single centre in Malaysia.
METHODS
This cross-sectional, descriptive, clinical study was conducted between 1 June 2014 and 31 December 2014 at University Malaya Medical Centre (UMMC), Kuala Lumpur, Malaysia. UMMC serves as a community hospital for children with thalassaemia who live in the vicinity. Ethical approval was obtained from the Medical Ethics Committee of UMMC (2013/1031.100). Written informed consent was obtained from all parents or guardians of children with TDT who took part in the present study.
A list of patients with TDT who were managed at the Paediatric Thalassaemia Daycare, UMMC, Malaysia, was retrieved from the paediatric thalassaemia database. All patients with TDT, diagnosed according to the Thalassemia International Federation’s guidelines (≥ 8 transfusions per year), were included. Patients who had a history of HSCT, concurrent autoimmune diseases or were newly diagnosed to have TDT and had received < 8 transfusions in the previous year were excluded from the study. Patient demographics, age at diagnosis, type of thalassaemia syndrome, transfusion history and history of iron chelation therapy were retrieved from patient medical records.
The various definitions and diagnostic criteria used for the diagnosis of endocrine dysfunctions in the study are shown in
Table I
Definition and diagnostic criteria of endocrinopathies used in the present study.
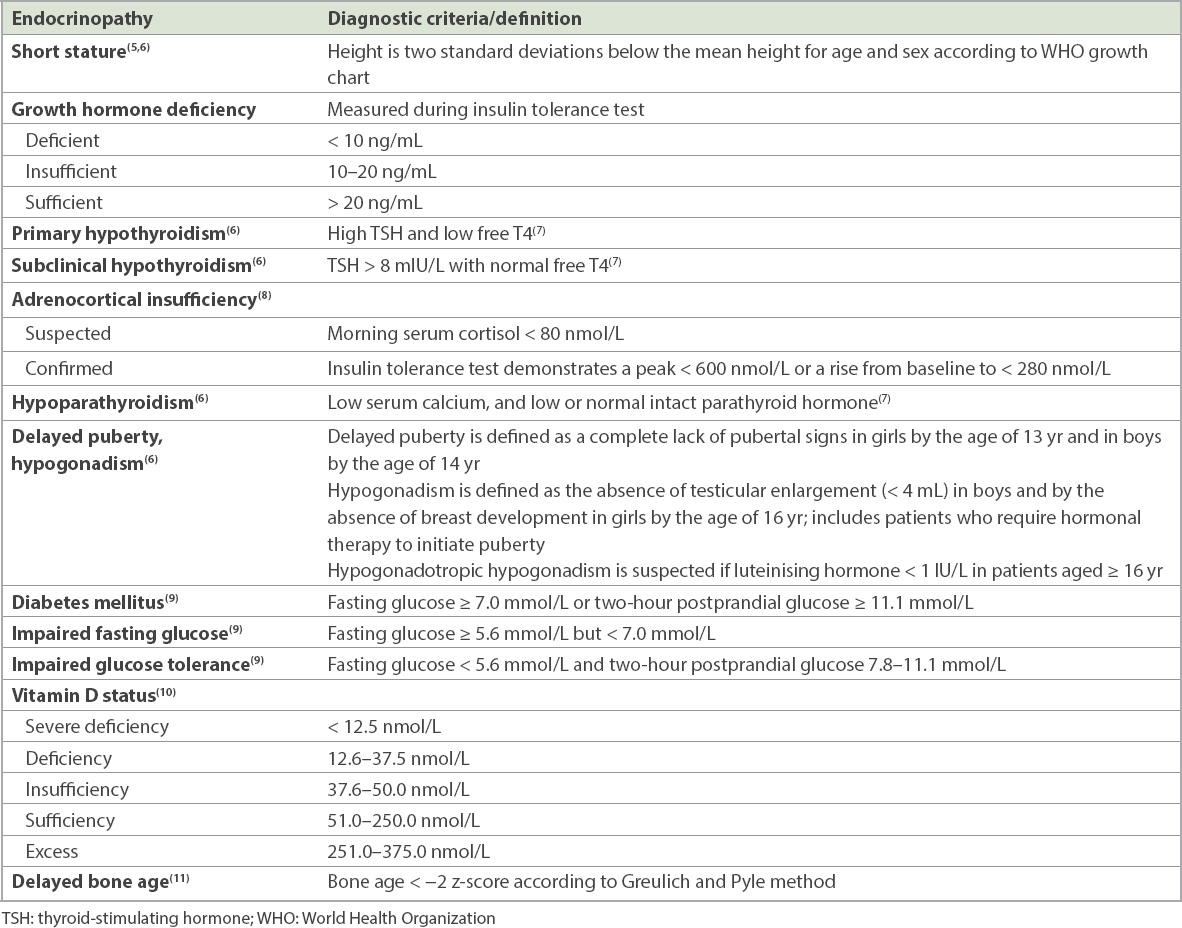
Complete blood count, serum ferritin, serum calcium, fasting plasma glucose, fructosamine, insulin-like growth factor-1, intact parathyroid hormone, 25-hydroxy vitamin D, thyroid-stimulating hormone, free T4, serum cortisol, luteinising hormone, follicle-stimulating hormone, estradiol in girls and testosterone in boys were all measured in the morning (~8 am) prior to blood transfusion. Oral glucose tolerance test (OGTT) was performed in all patients aged > 7 years. Growth hormone (GH) measurement was performed during the insulin tolerance test (ITT), and was indicated in patients with TDT who were well chelated but found to be short for their genetic potential (defined as height standard deviation [SD] score < 1.5 of the mid-parental height SD score).
Left hand and wrist radiography (anteroposterior view) was performed for bone age assessment. It was not performed in patients who had been assessed within the previous six months from the study period. Radiography was reported using the Greulich and Pyle method.(11) Average serum ferritin over the previous 18 months was also analysed. Ferritin had been monitored every six months, with a total of three readings in the 18-month period.
Data was managed using SPSS version 16.0 (SPSS Inc, Chicago, IL, USA). Median and range were reported for continuous variables and percentages were calculated for categorical variables. Univariate analysis of potential factors affecting the presence of endocrinopathy was conducted. Factors with a p-value < 0.5 were subjected to multivariate analysis. On multivariate analysis, factors with a p-value < 0.05 were considered to be statistically significant.
RESULTS
During the study period, a total of 95 patients with TDT were under the care of the Paediatric Thalassaemia Daycare service of UMMC. Of these, 13 patients who were excluded had the following characteristics: (a) non-transfusion-dependent thalassaemia (n = 2); (b) newly diagnosed (n = 5); (c) concurrent autoimmune disease (n = 2); and (d) previous HSCT (n = 4). Thus, a total of 82 patients were recruited for the present study.
The demographic features, red cell transfusion records and chelation regimens of these 82 patients are summarised in
Table II
Demographic characteristics, clinical features, red cell transfusion records and iron chelation regimens of children in the study.
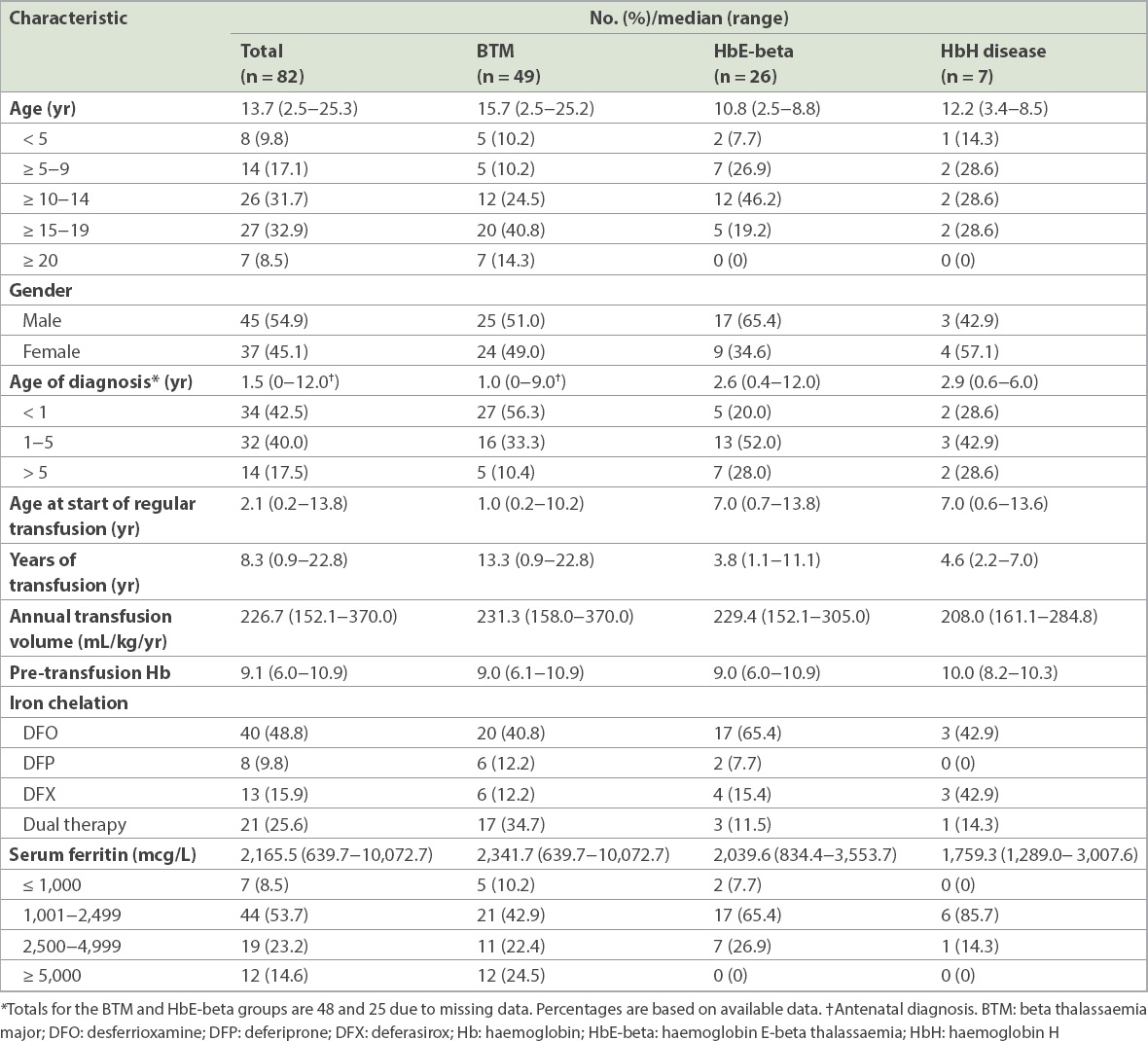
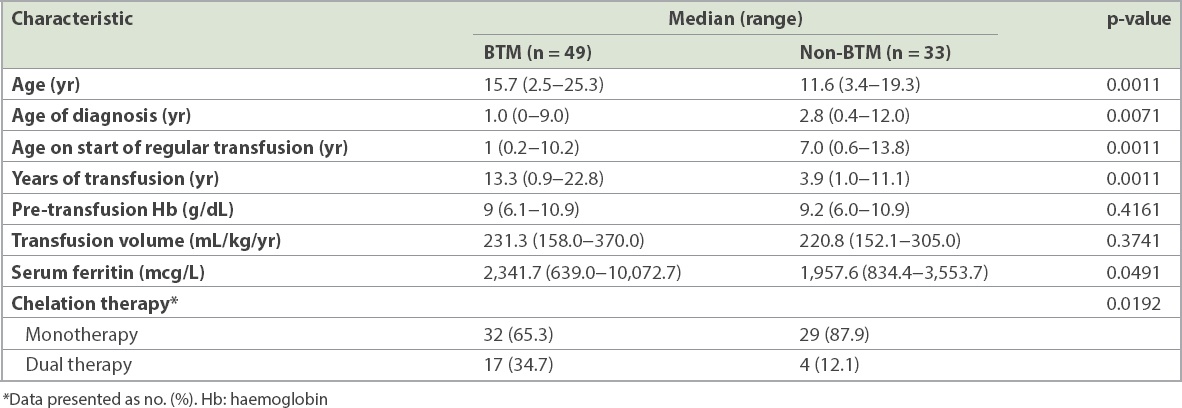
Table III
Clinical features of patients with beta-thalassaemia major (BTM) and non-BTM.
A total of 53 (64.6%) patients with a median age of 14.0 (range 2.5–25.3) years were found to have at least one endocrine disorder (
Table IV
Endocrinopathies in the study population.
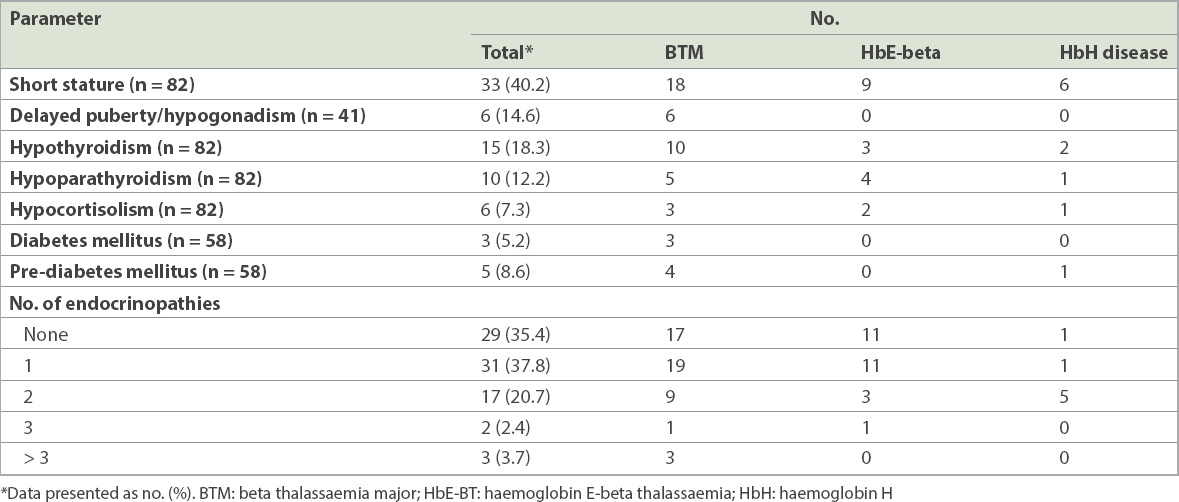
Table V
No. of endocrinopathies in children with transfusion-dependent thalassaemia according to age.
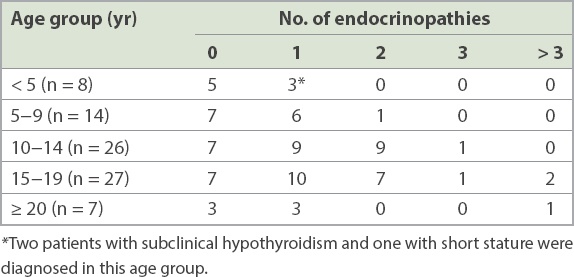
Short stature was the commonest endocrine dysfunction encountered. A total of 33 (40.2%) of the 82 patients had a height z-score ≤ –2 SD according to the World Health Organization growth chart. The median height z-score for the 82 patients was −1.75 (range –3.86 to 0.81). 27 (81.8%) of the 33 patients were aged 10−19 years. There was an almost equal number of male and female patients with short stature. In addition to short standing height, one patient also had truncal shortening (sitting height/standing height ratio z-score ≤ –2 SD). 10 (30.3%) of the 33 patients had ITT for assessment of short stature and nine were confirmed to have GH deficiency; all were commenced on recombinant human GH treatment.
Hypogonadism was another finding in the present cohort. There were 23 female patients aged ≥ 13 years and 18 male patients aged ≥ 14 years. Of these 41 patients, six female patients with BTM had delayed puberty/hypogonadism requiring hormonal replacement. Three of them were aged ≥ 16 years; one had primary ovarian failure (hypergonadotropic hypogonadism), while two had hypogonadotropic hypogonadism. In terms of hypothyroidism, 15 (18.3%) of the 82 patients, with a median age of 16.4 (range 2.6−20.1) years, had deranged thyroid function tests. Cases included subclinical hypothyroidism (n = 11) and overt hypothyroidism (n = 4) consisting of primary (n = 3) or secondary hypothyroidism (n = 1). The median age at diagnosis of hypothyroidism was 14.5 (range 12–20) years, while the median age at diagnosis of subclinical hypothyroidism was 15.7 (range 2.6–20.1) years. All patients with hypothyroidism had BTM, while 7 (63.6%) out of 11 patients with subclinical hypothyroidism had BTM.
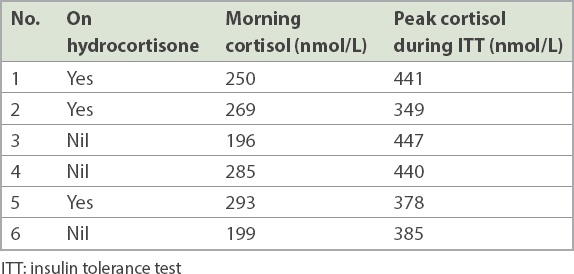
The median morning cortisol level was 249 (range 86–534) nmol/L. When a morning serum cortisol level of < 80 nmol/L was taken as the cut-off value to screen for adrenal insufficiency, none of the patients had a low serum cortisol level. However, six of the 10 patients who had ITT for the evaluation of short stature had hypocortisolism (
Table VI
Serum cortisol levels in six patients with hypocortisolism.
Hypoparathyroidism was diagnosed in 10 (12.2%) patients, who had a median age of 13.1 (range 6.8–25.3) years. Two were diagnosed prior to the present study, while another eight had hypocalcaemia (< 2.20 mmol/L) with a low-normal or low parathyroid hormone level, consistent with primary hypoparathyroidism. An additional seven patients had a normal serum calcium level but very low intact parathyroid hormone, suggesting subclinical hypoparathyroidism.
Among the 79 patients whose vitamin D level was measured, 17 (21.5%) had a low 25-hydroxy vitamin D level (< 50 nmol/L). Vitamin D was deficient in 8 (10.1%) patients and insufficient in another 9 (11.4%) patients. However, none had severe vitamin D deficiency. Two patients had concomitant hypoparathyroidism and vitamin D deficiency.
To test for impaired glucose tolerance, 58 patients (aged ≥ 7 years) underwent OGTT and 8 (13.8%) patients, who had a median age of 16.8 (range 11.4–20.1) years, were found to have abnormal glucose levels. 3 (5.2%) patients with BTM fulfilled the criteria for diabetes mellitus (DM). Another 4 (6.9%) patients had impaired glucose tolerance, while 1 (1.7%) patient had impaired fasting glucose. They were commenced on diet and lifestyle modifications and given advice on reducing their serum ferritin level.
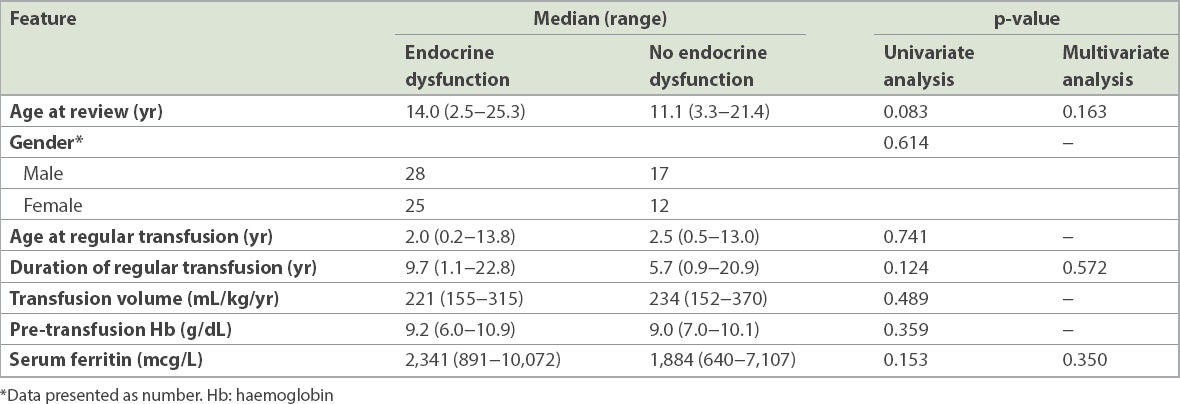
Various demographic and clinical factors were analysed to ascertain significant factors affecting the presence of endocrinopathy (
Table VII
Clinical features in children with transfusion-dependent thalassaemia, with and without endocrinopathy.
DISCUSSION
In the present study, about two-thirds of patients with TDT had at least one endocrine disorder. This was unacceptably high and occurred despite the availability of various iron chelation therapies. The majority of these patients had a serum ferritin level of ≥ 1,000 mcg/L, similar to those observed by Borgna-Pignatti et al.(12) The Thalassemia International Federation’s guidelines also recommend a serum ferritin level of < 1,000 mcg/L to prevent endocrine morbidities in patients with TDT.(6,13)
Short stature was the most prevalent endocrine disorder observed in the present study. The observed prevalence of 40.2% was much higher as compared to reports of 28.5%–30.8% from studies in developed countries,(3,14) but was relatively lower compared to similar figures of 55.0%–69.6% from developing countries.(15,16) Short stature in thalassaemia may be multifactorial. Potential causes include malnutrition, chronic anaemia, iron overload leading to GH deficiency, and genetic short stature. Patients who had optimum red cell transfusion and iron chelation therapy were selected for a provocation test as part of the management of short stature. Interestingly, nine out of 10 patients who had ITT for short stature were diagnosed to have GH deficiency. All of these patients received GH therapy. If ITT had been repeated in all patients with TDT and short stature, it is likely that more patients who had short stature due to GH deficiency would have been identified.
Hypogonadism is not uncommon in teenagers with TDT. However, it was only reported in 14.6% of our population. This result was lower as compared to other studies, with a reported prevalence of 40%–70%.(3,16) This may be due to the nature of the study design; patients recruited for the present study were much younger, and a significant number had not entered puberty. Nevertheless, it would be important to continue monitoring this group of children at the onset of puberty.
The prevalence of overt hypothyroidism observed in the present study was 4.9%.(3,14) However, the prevalence of subclinical hypothyroidism (13.4%) was much higher than in other studies, which noted a prevalence of 2.1%–8.9%.(14,16) Thus it is important to monitor the thyroid function of all patients with TDT, because the symptoms of hypothyroidism can be subtle.
Regarding hypocortisolism, its prevalence varied between centres, as the reference range of the morning cortisol level used was different in various studies. None of our patients were found to have hypocortisolism based on the morning serum cortisol value of < 80 nmol/L. However, of the ten patients with TDT who underwent a provocation test for growth evaluation (i.e. ITT), 6 (60%) had hypocortisolism. As the symptoms of hypocortisolism are non-specific and there is currently no acceptable recommended range for morning serum cortisol, a high index of suspicion and early referral to an endocrinologist is necessary. Serial morning serum cortisol measurement may be a good start to identify early hypocortisolism, to be followed by a short Synacthen test.(8)
The prevalence of DM in this study was 5.2%. OGTT was arranged for all patients aged older than seven years. Since none of the patients were symptomatic, a repeat OGTT in the near future would be needed to establish a definite diagnosis of DM. In addition, 8.6% of the study population was diagnosed as having pre-DM (i.e. impaired glucose tolerance or impaired fasting glucose). Similar findings were also noted from other centres, where the prevalence of pre-DM was higher than that of DM.(16,17) For this group of patients, it is important to recommend strict iron chelation therapy to reduce their serum ferritin level, lifestyle improvement through diet control and increasing physical activity to avoid progression to overt DM.
The prevalence of hypoparathyroidism in the present study was 12.2%, which is considerably higher than the 1.4% observed by Vogiatzi et al.(14) This could be attributed to the difference in the definition of hypoparathyroidism. In most other studies, a combination of low calcium, raised serum phosphate and alkaline phosphatase levels was used as the definition of hypoparathyroidism.(14) In the present study, a combination of low serum calcium level and the presence of low or normal intact parathyroid hormone was used. We also noticed a significant number of patients who had subclinical hypoparathyroidism. Thus, it is essential to monitor the serum calcium level of these patients carefully.
There were several limitations to the present study. As it was a cross-sectional review, we were unable to assess the dynamic changes of endocrinopathies seen in patients with TDT, particularly pubertal arrest and secondary hypogonadism. Second, parental heights were unavailable, and hence we were unable to exclude genetic causes of patients’ short stature. It was also a single-centre study and the sample size was relatively small. Third, average serum ferritin was taken as the sole marker for iron overload. An average of the latest 18-month serum ferritin measurements used in the present study could not fully reflect the long-term impact of haemochromatosis on endocrinopathy. Furthermore, patients’ adherence to iron chelation therapy was not addressed in the present study.
In conclusion, the present study demonstrates that endocrinopathy is a major health issue encountered by patients with TDT despite the standard medical therapies. Close monitoring and timely institution of hormonal therapy are essential to improve their endocrine outcome and general well-being.
ACKNOWLEDGEMENT
The present study was funded by the Postgraduate Research Fund, Cycle 2/2013 (GL: H-00000-60004-E13110) and Iron Chelation Fund of University Malaya, Kuala Lumpur, Malaysia.