

Abstract
INTRODUCTION
Fragile X syndrome (FXS) is the most prevalent X-linked intellectual disability (ID) and a leading genetic cause of autism, characterised by cognitive and behavioural impairments. The hyperexpansion of a CGG repeat in the fragile X mental retardation 1 (FMR1) gene leads to abnormal hypermethylation, resulting in the lack or absence of its protein. Tools for establishing the diagnosis of FXS have been extensively developed, including assays based on triplet-primed polymerase chain reaction (TP-PCR) for detection and quantification of the CGG trinucleotide repeat expansion, as well as determination of the methylation status of the alleles. This study aimed to utilise a simple, quick and affordable method for high sensitivity and specificity screening and diagnosis of FXS in institutionalised individuals with ID.
METHODS
A total of 109 institutionalised individuals at the Center for Social Rehabilitation of Intellectual Disability Kartini, Temanggung, Central Java, Indonesia, were screened in a three-step process using FastFrax™ Identification, Sizing and Methylation Status Kits.
RESULTS
Two samples that were classified as indeterminate with respect to the 41-repeat control at the identification step were subsequently determined to be non-expanded by both sizing and methylation status analyses. Two samples classified as expanded at the identification step were determined to carry full mutation expansions > 200 repeats that were fully methylated using sizing and methylation status analyses, respectively, yielding a disease prevalence of 1.83%.
CONCLUSION
Repeat expansion and methylation-specific TP-PCR is practical, effective and inexpensive for the diagnosis of FXS, especially in high-risk populations of individuals with ID of undetermined aetiology.
INTRODUCTION
Fragile X syndrome (FXS) is the most prevalent inherited cause of intellectual disability (ID). It is caused by hyperexpansion of a CGG trinucleotide repeat in the 5’ untranslated region of the fragile X mental retardation 1 (FMR1) gene to more than 200 repeats, which is referred to as a full mutation (FM). Concomitant hypermethylation of the repeat expansion silences expression of FMR1, resulting in the lack or absence of fragile X mental retardation protein.(1,2) The CGG repeat in normal (NL) individuals ranges from 5 to 45 repeats. Alleles with 45–54 repeats are classified as intermediate (IM) or ‘grey zone’, while alleles of 55–200 repeats are classified as premutation (PM). FXS occurs in approximately one in 4,000 males and one in 6,000 females in the general population, and in 1.7% of the intellectually disabled population in Indonesia.(3,4)
FXS is characterised by cognitive, social, emotional and behavioural impairment. Adaptive functioning impairment in individuals with FXS varies from moderate to severe, and is correlated with the degree of hypermethylation of the FMR1 gene.(5) Carriers of the FMR1 PM allele were previously presumed to be clinically normal. However, more recent studies have found that PM carriers are at risk of fragile X-associated phenotypes. Female carriers have a higher risk of fragile X-associated premature ovarian insufficiency, while 30% of male PM carriers exhibit fragile X-associated tremor/ataxia syndrome, where the patient shows signs of intention tremor from the age of 50, which gradually progresses to ataxia.(6)
Behavioural phenotypes that may be found in FXS include social anxiety and social avoidance, gaze avoidance, inattention, social deficits, tactile defensiveness, hyperactivity and impulsivity. These traits can also be found in autism spectrum disorders (ASD), and 30% of patients with ASD actually have FXS.(7,8) However, some autistic symptoms that present in individuals with FXS do not meet the diagnostic criteria of ASD. In the Indonesian context, individuals with ID are mostly assigned to special education schools or institutions. However, individuals with mild behavioural impairment are more likely to be institutionalised than those with more severe conditions.
Many tools have recently emerged for establishing the diagnosis of ID, including FXS. Although Southern blot is a gold standard DNA methodology for FXS diagnosis, there are now more advanced methods that are simple, rapid and accurate. A closed-tube triplet-primed polymerase chain reaction (TP-PCR) and melting curve analysis (MCA) assay is one attractive tool for rapid FXS screening. The FastFrax™ Identification Kit and Sizing Kit enable rapid detection and sizing of FMR1 CGG repeat expansion mutations, respectively.(9) In addition, analysis of FMR1 allele methylation status is also now more affordable and practical.(10) Screening of repeat expansion and methylation status in individuals with FXS is important, as it provides a definitive diagnosis for the family and/or caregiver, and facilitates genetic counselling of the affected individual and relatives by the healthcare provider. Screening for FXS in high-risk populations such as those with ID is an effective means of identifying affected individuals and determining the prevalence of ID, and allows family members to undergo post-test genetic counselling sessions and cascade testing.(11) This study utilised a simple, quick and inexpensive method with high sensitivity and specificity for diagnosing FXS among institutionalised individuals with ID.(12)
METHODS
All participants underwent physical, dysmorphological and behavioural examinations by experienced physicians. Those who had clinical presentations of Down syndrome were excluded. The Hagerman fragile X checklist was used for all individuals who matched inclusion criteria.(7) A total of 109 individuals from the Centre for Social Rehabilitation of Intellectual Disability Kartini, Temanggung, Central Java province, Indonesia, were included and subjected to FMR1 molecular analysis using the three FastFrax FMR1 Identification, Sizing and Methylation Status Kits (The Biofactory Pte Ltd, Singapore). The study was performed in three stages, starting with identification of expansion-positive individuals, followed by sizing of the CGG repeats and, subsequently, determination of FMR1 methylation status. This study was approved by the Ethics Committee of the Medical Faculty, Diponegoro University/Kariadi Hospital, Semarang, Central Java, Indonesia. Prior to subject enrolment, informed consent was obtained from the participants’ parents or guardians.
To identify CGG repeat expansion, the FastFrax FMR1 Identification Kit was used. This involved TP-PCR, followed automatically by MCA in a closed-tube reaction. The TP-PCR assay was performed according to the manufacturer’s instructions for use in 25-mL volumes with 50 ng genomic DNA for each test. PCR amplification and subsequent MCA were performed on a RotorGene Q high resolution melting (HRM) instrument (Qiagen, Valencia, CA, USA). Thermal cycling comprised an initial denaturation step at 95°C for 15 minutes, followed by 40 cycles of 99°C for two minutes, 65°C for two minutes and 72°C for three minutes, ending with a final extension at 72°C for 10 minutes. MCA involved a denaturation step at 95°C for one minute and a temperature ramp from 75°C to 99°C at a rate of 0.5°C, with a five-second hold at each step.(9)
Data interpretation at the identification stage involved an initial visual check for aberrant results (e.g. significant lower fluorescent signals) compared to the controls, followed by determination of the temperature at which the sample’s −dF/dT (negative first derivative of fluorescence against temperature) value drops to baseline. Each sample was classified as non-expanded or expanded depending on whether its resumed baseline –dF/dT temperature was lower or higher than that of the selected threshold control sample.(9) Three DNA samples with well-characterised FMR1 CGG repeats of 30, 41 and 53 CGG repeats (NA06890, NA20244, NA20230) (Coriell Cell Repositories, Camden, NJ, USA) were used as controls to establish threshold/cut-off resumed baseline –dF/dT temperatures.
Sizing analysis was performed in all 109 samples using the FastFrax FMR1 Sizing Kit, which involved performing a TP-PCR reaction followed by capillary electrophoresis (CE) analysis of an aliquot of the TP-PCR product. A female FM DNA sample (NA07537, Coriell Cell Repositories) was used as a reference positive control sample. The TP-PCR assay was performed according to the manufacturer’s instructions for use in 15-mL volumes, using 100 ng of genomic DNA for each test. PCR was performed using the Bio-Rad C1000 thermal cycler (Bio-Rad Laboratories, Hercules, CA, USA). Thermal cycling comprised an initial denaturation at 95°C for 15 minutes, followed by 40 cycles of 99°C for 45 seconds, 55°C for 45 seconds, and 70°C for eight minutes with an extension of 15 seconds at each cycle, ending with a final extension at 72°C for ten minutes.(12)
4 μL of each amplicon was mixed with 0.5 μL of MapMarker 1000-ROX (Bioventures Inc, Murfreesboro, TN, USA) and 9 μL of Hi-Di Formamide (Applied Biosystems, Forster City, CA, USA). Sample mixtures were denatured at 95°C for five minutes, rapidly cooled to 4°C on a thermocycler and subjected to CE through a 50-cm capillary loaded with POP-7 polymer in an ABI 3730 xL DNA analyser (Applied Biosystems). The machine was set to an injection voltage of 1.2 kV for 18 seconds, followed by CE at 15 kV for 50 minutes.(12)
CE results were analysed using PeakScanner™ 2 software (Applied Biosystems). The number of CGG repeats was determined by either one of two approaches: peak counting (for shorter alleles of the NL or small PM size range) or approximation using peak distance in base pairs following the manufacturer’s instructions. Samples were classified into NL, IM, PM and FM genotypes based on the reported repeat size of the largest allele.(12) The sizing kit enabled quantification of the exact sizes of CGG repeats up to 200 and the reporting of FM samples that had > 200 repeats.
Methylation status was determined as expanded or indeterminate relative to the 41-repeat cut-off control for all samples that were classified at the identification stage, as well as a few selected samples classified as normal for comparison purposes.
Genomic DNA was treated with sodium bisulfite using the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions for use. Analysis was performed on sodium bisulfite-modified genomic DNAs using the FastFrax FMR1 Methylation Status Kit, which involves a duplex TP-PCR reaction in a single tube to detect and differentiate between methylated and unmethylated FMR1 alleles using CE. A sodium bisulfite-modified positive control sample, an unmodified DNA control sample and a no-template control were included in the analysis. Methylation status was determined by analysing the CE results in the blue fluorescent dye (fluorescein amidite) channel for the unmethylated allele, and the green fluorescent dye (hexachloro-fluorescein) channel for methylated alleles.
RESULTS
All DNA samples were first screened using the FastFrax FMR1 Identification Kit. Three well-characterised Coriell cell line DNA samples carrying 30, 41 and 53 CGG repeats, respectively, were used to generate resumed baseline –dF/dT temperatures. This enabled classification of the 109 test samples as expanded or non-expanded with respect to the respective controls (
Fig. 1
Chart shows the number of samples clarified as non-expanded, expanded and indeterminate in relation to the respective controls. Rpt: repeats
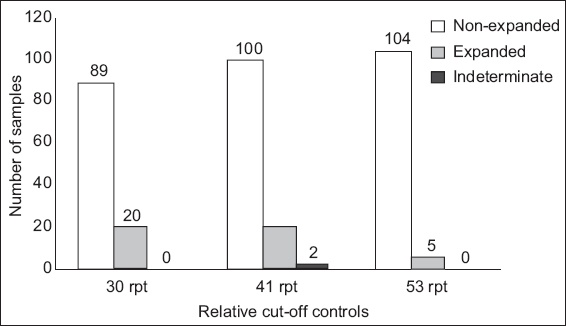
Of the five samples classified as expanded in relation to the 53-repeat control by the identification kit, two samples were found to carry an FM allele (> 200 repeats) using the sizing kit. One was an FM male sample, while the other was a female sample that was heterozygous for a normal allele of 29 repeats and an FM allele of > 200 repeats. Three samples (TG-153, TG-174 and TG-192) displayed resumed baseline –dF/dT temperatures that overlapped with the 53-repeat control, although their melt peak curves dipped towards baseline earlier than the 53-repeat control, thus making the results difficult to interpret (
Fig. 2
Graph shows melt profiles for three indeterminate samples, TG-153, TG-174 and TG-192. All curves dipped towards baseline earlier compared to the 53-repeat control. Samples TG-148 and TG-165 were indeterminate to the 41-repeat control, and sample TG-140 was not expanded.
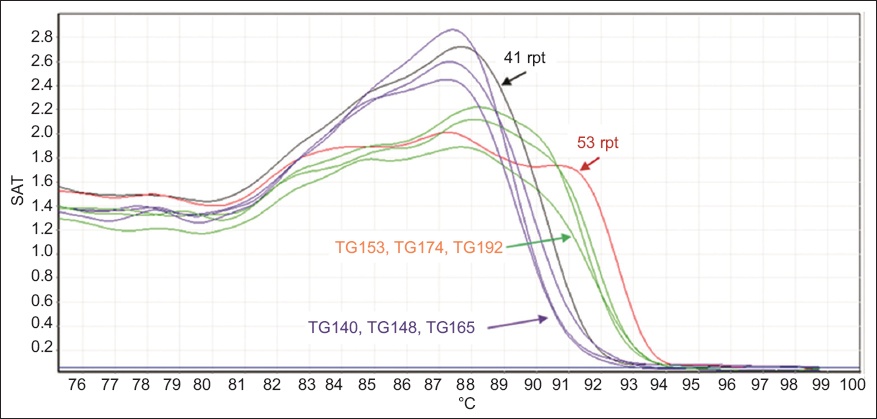
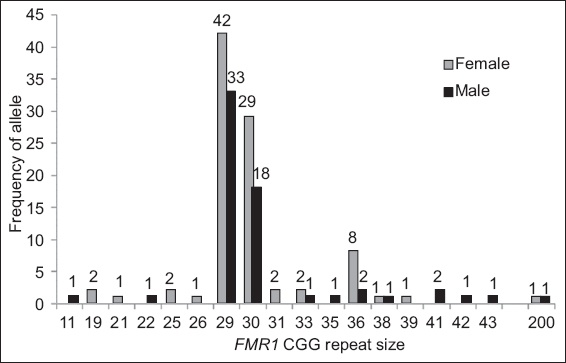
Fig. 3
Chart shows frequency of CGG repeat lengths in the test cohort of 109 participants (46 female, 63 male).
Methylation status analysis was performed on five samples that were classified by the identification kit as expanded in relation to the 53-repeat control, together with six non-expanded samples that were included for comparison purposes (
Table I
Results of the 11 samples tested using the methylation status kit.
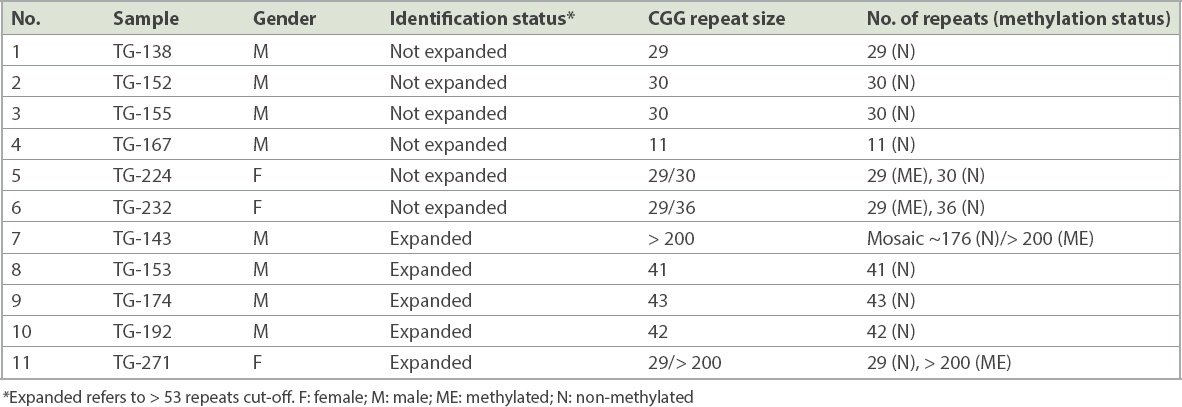
Fig. 4
Charts show capillary electrophoresis profiles of the male sample (TG-143, Subject 1) carrying a full mutation allele, as determined by the sizing kit. (a) The FAM (fluorescein amidite) channel shows a unmethylated allele with 176 CGG repeats, while (b) the HEX (hexachloro-fluorescein) channel shows a fully methylated allele with > 200 CGG repeats. rpt: repeats
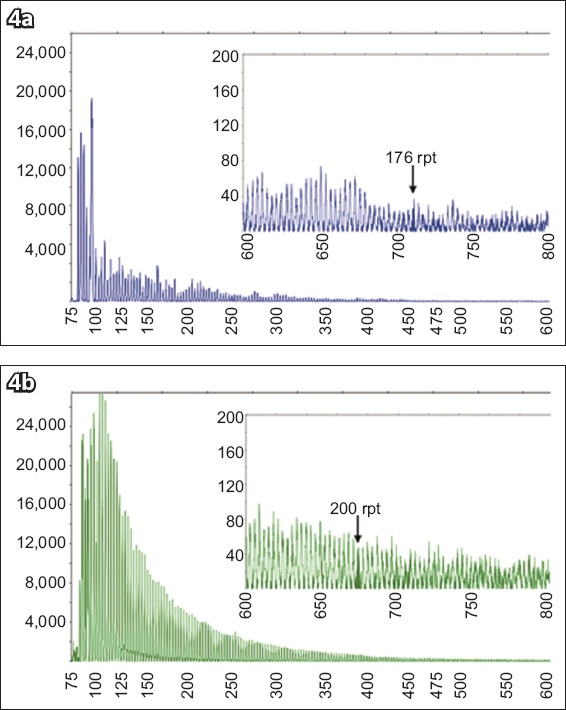
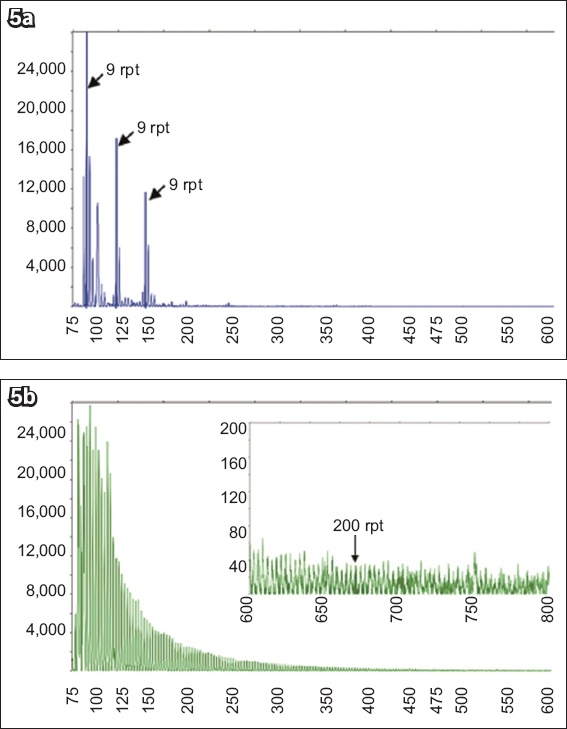
Fig. 5
Charts show capillary electrophoresis profiles of the female sample (TG-271, Subject 2), carrying a full mutation allele, as determined by the sizing kit. (a) The FAM (fluorescein amidite) channel shows a unmethylated allele with 29 CGG repeats, including two AAG interruptions, while (b) the HEX (hexachloro-fluorescein) channel shows a fully methylated allele with > 200 CGG repeats. rpt: repeats
Subject 1 was a 25-year-old male participant with moderate ID and mild behaviour who possessed some physical characteristics commonly found in individuals with FXS, such as a long face, large and prominent ears, macro-orchidism and hyperextensible joints. In contrast, Subject 2 was a 22-year-old female participant with mild ID and without any obvious FXS physical features. The Hagerman checklist was performed prior to molecular analysis and resulted in low scores of 13 for Subject 1 and 5 for Subject 2 (threshold score ≥ 16).(7)
DISCUSSION
In this study, repeat expansion and methylation-sensitive TP-PCR and MCA were utilised as a simple method to identify FMR1 trinucleotide repeat expansions, which are responsible for FXS among intellectually disabled individuals. Conventional analysis such as fragile site expression and Southern blotting are known to be laborious, despite the specificity and standardised workups. Analysis of FMR1 expansion mutations using three-step PCR-HRM is less time-consuming while maintaining its specificity and sensitivity in CGG trinucleotide repeat expansion. Screening and quantification of CGG trinucleotide number have yielded results of 100% sensitivity (95% confidence interval [CI] 91.0%–100%) and 99.6% specificity (95% CI 98.5%–99.9%) in detecting expansions of > 55 CGG repeats that have 100% sensitivity (95% CI 91.03%–100%) and 100% specificity (95% CI 97.64%–100%) in categorising patient samples into NL, IM, PM and FM genotypes.(9,12) In addition to these two steps, methylation status step analysis provides additional information on X-inactivation skewing and mosaicism in FM individuals, which may help to predict phenotypic severity in affected individuals.
The most prevalent CGG repeat length in this study was 29 repeats, followed by 30 and 36 repeats. These findings are consistent with those of Faradz et al, who concluded that the 29 and 30 CGG repeat alleles were the most prevalent in the Indonesian and Asian populations, with 29 repeats being more common than 30.(6,13) Another study conducted in 2012 by Winarni et al showed that the most common CGG repeat allele in an Indonesian high-risk population was 30 repeats, followed by 29 repeats, with a smaller percentage of 35 and 36 repeats.(11)
Using sizing analysis, two out of five samples that were classified as expanded with respect to the 53-repeat reference control (TG-143 and TG-271) were determined to carry FM alleles of > 200 repeats, although the exact sizes of the FM expansions could not be determined by the kit. The remaining three samples that were classified as expanded (TG-153, TG-174 and TG-192) were false positives. At the time of testing, the decision was taken to err on the side of caution with respect to sample classification, despite the fact that these samples were clearly distinct among other samples. This conservative interpretation approach ensures 100% sensitivity in detecting expansion mutations while contributing to a low but acceptable false positive rate in large cohort screening situations. In this study, all samples that underwent identification analysis were also subjected to sizing analysis, confirming that the samples classified as negative by the identification analysis were truly negative. By subjecting to sizing analysis only those samples that were classified as positive by identification analysis, we were able to rule out false positive results in this prospective analysis.
Further analysis using the methylation status kit confirmed the FM expansions and showed that they were aberrantly hypermethylated in both individuals, consistent with their FXS phenotype.(9) The finding of mosaic alleles in the male subject (Subject 1) is consistent with his previously observed low fragile X checklist score and mild behaviour impairment, and thus clinical diagnosis should be combined with molecular screening to improve the accuracy of FXS screening.
FXS screening plays an important role in facilitating further cascade testing of the family members of affected individuals. FXS testing rates remain low in most populations worldwide, since the physical characteristics are usually unremarkable, making it an indistinguishable syndrome.(14) FMR1 screening involving TP-PCR, followed automatically by MCA in a closed-tube reaction (FastFrax), was less time-consuming than conventional PCR followed by Southern blot analysis, has very high sensitivity and can be applied in Indonesia. Stepwise analysis from identification, sizing and methylation status in this study demonstrates the relevance of screening in a previously unknown population. The availability of screening will increase earlier identification of individuals at high risk for developmental delay and ID, as well as avoiding a diagnostic odyssey, as explained in some recent studies.(15-17) The screening approach using an identification kit removes the need for additional CE with non-expanded samples, hence providing cost savings. The combined approach for this study demonstrated that conservatively, < 15% of samples needed to be characterised using the conventional method, resulting in cost savings of at least 30%. The savings could be greater when such a study is applied in a much larger population with lower prevalence (e.g. newborn screening).
There have been advances in research on therapeutic regimens focusing on the molecular pathophysiology of FXS. Insulin and insulin-like growth factor pathway, serotonin pathway and endocannabinoid pathway are some of the therapeutic targets in which preclinical trials have obtained promising results.(18) The increased diagnostic rates in FXS need to be coexistent with the development of new treatments for the benefit of patients and their families.
The repeat expansion and methylation-sensitive TP-PCR methods using FastFrax kits represent practical methods for high specificity and sensitivity FXS screening and diagnosis. FMR1 screening of high-risk populations such as those with ID may result in identification of significant numbers of otherwise undetected FXS individuals. Additionally, recent studies suggested that therapeutic strategies for FXS will be available in the near future, making prompt diagnosis important for management and improvement of FXS in individuals.
About the First Author

Dr Nydia Sihombing is a medical doctor and human genetics researcher at the Center for Biomedical Research (CEBIOR), Faculty of Medicine, Diponegoro University, Indonesia. Her research topics are mainly on the genetics of intellectual disability and multiple congenital anomalies, specifically fragile X syndrome and other syndromic intellectual disabilities.
ACKNOWLEDGEMENTS
This study was supported by funds from the Ministry of Research, Technology and Higher Education of Indonesia (PUPT Kemenristek Dikti Grant no. 176-13/UN7.5.1/PG/2016 and PMDSU Grant no. 345-11/UN7.5.1/PG/2017). We would like to thank the Balai Besar Rehabilitasi Sosial Bina Grahita Temanggung institution, patients and caregivers for participating in our study. We would also like to thank Elsa Gusrianti, Suharta and Mifta Mahardika (Faculty of Medicine, Diponegoro University, Indonesia) for data collection, and to Yeo Minli (BioFactory, Singapore) and Lusi Suwarsi (Center for Biomedical Research, Faculty of Medicine, Diponegoro University, Indonesia) for laboratory technical assistance.