

Abstract
Subacute sclerosing panencephalitis (SSPE) is a rare, slowly progressing but invariably fatal disease that is related to a prior measles virus infection and most commonly affects paediatric patients. Magnetic resonance (MR) imaging is the modality of choice for determining such changes in white matter. SSPE typically demonstrates bilateral but asymmetric periventricular and subcortical white matter involvement. We herein report a rare case of unilateral white matter involvement in a 13-year-old boy with SSPE that closely simulated Rasmussen’s encephalitis. To the best of our knowledge, this is the first report of an atypical presentation on MR imaging in which SSPE was a rare cause of unilateral brain parenchymal involvement in a patient with intractable seizures.
INTRODUCTION
Subacute sclerosing panencephalitis (SSPE) is a rare, chronic, slowly progressive, debilitating and slow virus infection. It typically affects children and young adults, and is related to a prior measles infection in childhood. The illness begins as behavioural disturbances, followed by myoclonic jerks, before ultimately resulting in dementia and a vegetative state. Diagnosis is usually made based on clinical history, typical electroencephalogram (EEG) changes and measles titres in cerebrospinal fluid (CSF) and serum. Magnetic resonance (MR) imaging plays a supportive role in its diagnosis and helps to exclude other causes. SSPE commonly presents with bilateral asymmetrical periventricular and subcortical white matter hyperintensities.(1,2) To our knowledge, unilateral cerebral involvement on MRI has not been previously described in patients with SSPE. We herein present a rare patient with SSPE, in whom unilateral brain parenchymal involvement mimicking Rasmussen’s encephalitis was seen.
CASE REPORT
A 13-year-old boy presented to the paediatric department at Christian Medical College and Hospital, Vellore, Tamil Nadu, India, with a three-year history of seizures associated with jerking movements of the upper arms. He also had a two-year history of weakness in all four limbs and loss of comprehension and reciprocation. No past history of febrile rash or other similar complaints in the family were noted. On examination, the patient was bedridden, stuporous and not oriented to time, place or person. His Glasgow Coma Scale score was 6/15 (E4V1M1). There were no cranial nerve deficits. Generalised hypotonia with grade 0 power in all four limbs and myoclonic jerks were observed. No tremors, choreoathetosis, tics or fasciculation were noted. Laboratory investigations indicated that blood sugar levels, blood counts, electrolyte levels, liver function test and urinalysis were normal. A workup for inborn errors of metabolism was negative.
MR imaging was performed and showed subtle unilateral cortical hyperintensity, and subcortical and periventricular white matter hyperintensities involving most of the right cerebral hemisphere, with relative sparing of the frontal lobe (
Fig. 1
T2-W axial MR images show (a) subtle cortical hyperintensity (arrows) involving the right temporo-occipital lobes; (b) subcortical white matter hyperintensity in the parietal lobe (arrows); and (c) diffuse deep and periventricular white matter hyperintensity (arrows) in the right cerebral hemisphere.
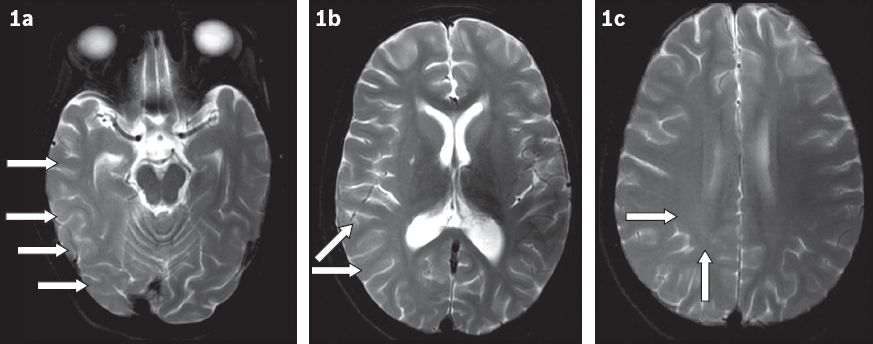
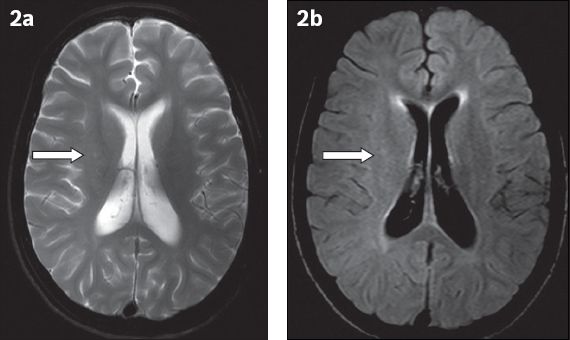
Fig. 2
(a) T2-W and (b) fluid-attenuated inversion recovery axial MR images show subtle putaminal hyperintensity (arrows) on the right side when compared to the contralateral normal side.
Based on these imaging findings, an initial diagnosis of Rasmussen’s encephalitis was made, with SSPE also being considered in the differential diagnosis due to the patient’s clinical history of myoclonic jerks. A subsequent EEG showed a burst suppression pattern suggestive of SSPE. CSF and serum were collected concomitantly and tested for the measles antibody titre, which was found to be significantly elevated. The clinical history, laboratory findings and EEG changes were all in keeping with SSPE.
DISCUSSION
SSPE is an invariably fatal, chronic, debilitating and slow viral infection. It usually affects children and adolescents aged 5–15 years.(1,2) The disease manifests about six years after measles infection.(1) It is presumed to be caused by a reactivation of a latent measles infection.(1) The long latency and slow evolution of the disease are partly explained by the failure to produce antibodies specific to the virus proteins. SSPE is clinically characterised by the insidious onset of behavioural changes, subsequently followed by myoclonic jerks and seizures that finally result in dementia and a comatose state.
MR imaging is the imaging modality of choice for determining white matter changes in patients with SSPE, as the disease is known for its specific propensity to involve periventricular and subcortical white matter in the parietal region. Involved areas appear as hyperintensities on T2-weighted MR imaging and fluid-attenuated inversion recovery images.(2) Contrast enhancement is usually absent. In the early stages of the disease, there is cortical and subcortical white matter involvement, with mild mass affect, which further progresses to involve the periventricular white matter, finally resulting in atrophic changes.(2) The affected white matter shows facilitated diffusion, with high apparent diffusion coefficient (ADC) values. The ADC values may help in the early diagnosis of the disease in patients for whom conventional MR imaging findings are normal.(3) Other regions that are less commonly involved include the basal ganglia, brainstem and thalamus.(2)
Asymmetrical white matter involvement is a well-described MR imaging feature of SSPE.(2) However, to the authors’ knowledge, complete unilateral involvement has not been previously described in the literature. Unilateral white matter involvement on MR imaging in patients with SSPE poses a diagnostic dilemma for clinicians, as in the case of our patient, as Rasmussen’s encephalitis would also be a likely diagnosis. However, the clinical history of myoclonic jerks was indicative of SSPE in our patient.
Kornberg et al have previously reported biopsy-proven, focal mass-like infiltrative involvement of the cerebral hemisphere in a 16-year-old girl with SSPE who presented with intractable seizures.(4) Similarly, Shivji et al reported asymmetric, predominantly left-sided, cerebral hemispheric involvement on MR imaging in a 12-year-old boy with SSPE.(5) These reports and the findings in our patient highlight the importance of clinicians remaining aware of the possibility of SSPE, apart from Rasmussen’s encephalitis, being a cause of unilateral brain parenchymal hyperintensity on MR imaging in paediatric or adolescent patients presenting with intractable seizures.
On imaging studies, Rasmussen’s encephalitis characteristically shows unilateral involvement of the frontal and temporal regions with cortical swelling in the initial stages, followed by hemiatrophy.(6) In contrast, unilateral involvement is an extremely rare imaging finding in patients with SSPE. Similarly, involvement of the basal ganglia is more commonly seen in Rasmussen’s encephalitis compared to SSPE.(7) However, our study suggests that imaging findings alone cannot differentiate SSPE from Rasmussen’s encephalitis, as only unilateral cerebral involvement was found in our patient. A definitive diagnosis in such patients would therefore require a correlation of clinical findings, the results of EEG and CSF analyses, and imaging studies.
Clinical history can be of help in the differential diagnosis of such patients, as Rasmussen’s encephalitis presents with a history of intractable seizures, with a typical pattern of epilepsia partialis continua, which is characterised by clonic movements of the face and upper extremities.(8) On the other hand, SSPE is characterised by myoclonic jerks, with a past history of measles. Typical EEG changes in SSPE include periodic bursts of bilateral, sharp and slow wave complexes with the suppression of electrical activity. The presence of measles antibodies in the serum and CSF of patients is also a useful point of reference for establishing the final diagnosis.(1)
To conclude, SSPE, much like Rasmussen’s encephalitis, can rarely present as unilateral brain parenchymal involvement on MR imaging studies. Clinicians should remain alert to the variations possible in the presenting features of these lesions. Detailed workups involving the patient’s clinical history, laboratory investigations and neurophysiological and imaging studies should be carried out to arrive at a definitive diagnosis in these patients.