

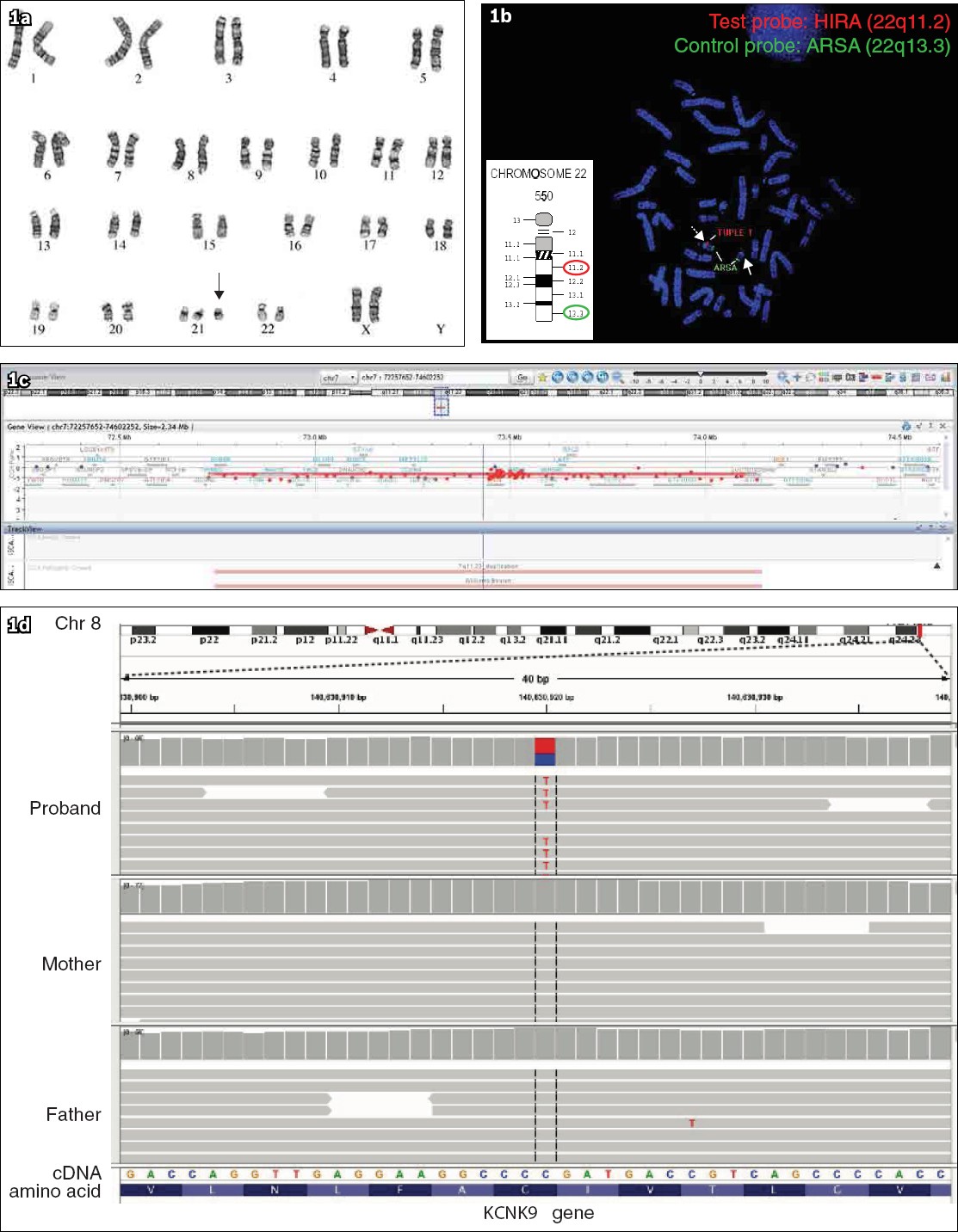
Abstract
Genomic medicine entails the use of an individual’s genomic information in his or her clinical care to aid diagnosis and personalise management. As genetic conditions significantly contribute to neonatal and paediatric morbidity and mortality, paediatricians can incorporate genomic medicine in their clinical practice. Through this review, we aim to describe the basic concepts of genomic medicine, discuss the types of genetic and genomic testing available, present examples of how genomics is already being used in paediatrics and summarise the challenges of genomic medicine. The purpose of this review is to help paediatricians decide the suitable genetic or genomic test, understand the possible outcomes of testing and appreciate the requirements of appropriate counselling of patients.
INTRODUCTION
The Human Genome Project, completed in 2003, resulted in the generation of the first draft of the human DNA sequence. This has prompted numerous initiatives aimed towards personalised medicine that is tailored to an individual’s genetic make-up. Options for genetic testing have increased exponentially with technological advances that allow rapid, high-throughput DNA sequencing. While traditional cytogenetic tests, such as karyotyping and fluorescence in situ hybridisation, require cell cultures to visualise numerical or structural abnormalities of chromosomes, molecular genetic tests can be performed on DNA extracted from nucleated cells, such as fresh blood, saliva or even stored tissue sample. These molecular genetic tests include chromosomal microarray analysis (CMA), single-gene- or gene-panel-targeted variant testing, sequencing of whole genes or panels of genes, and sequencing of the entire coding and noncoding regions of the human genome. Knowledge and understanding of the wide array of genetic tests, especially their clinical applications, is essential for the paediatrician, given that most genetic disorders present during infancy and childhood. In fact, congenital anomalies are identified in approximately 3% to 6% of live births,(1) and they contribute significantly to neonatal and infant hospital admissions and mortality.(1-3) In this review, we present the new advances in genomic technology and discuss the application of genomic tests in paediatric practice. In addition, we discuss the potential challenges in the realisation of precision medicine for better personalised patient-centric care in the near future.
WHAT IS GENOMIC MEDICINE?
A genome is the complete DNA sequence of an organism and includes genes as well as the intergenic regions. In humans, this includes the approximately 20,000 genes present in our entire genome. The term ‘genomics’ refers to the study of an organism’s genome, whereas genetics refers to the analysis of single genes and their inheritance. Genomic medicine is defined as the practice of using an individual’s genomic information in his or her clinical care.(4) It is increasingly applied in the fields of rare and undiagnosed diseases, oncology, pharmacology and infectious diseases. The integration of genomics into clinical care is made possible with increased understanding of the role of genomic information in human health and diseases through a broad range of collaborative research and improved technologies and computation analysis in interpreting this information. In the realm of rare paediatric diseases, accurate genetic diagnosis allows access to relevant information in the literature, targeted management and surveillance for the patient, and better understanding of the prognosis and genetic implications to the patient’s existing and future family members.(5-7)
TYPES OF GENETIC AND GENOMIC TESTS
Karyotyping (chromosome analysis)
Karyotyping is a cytogenetic method that involves the analysis of numerical and structural chromosomal abnormalities under a light microscope.(8) In this method, circulating lymphocytes from peripheral blood or other cell types from the skin, bone marrow, chorionic villi or cells from amniotic fluid are cultured under strict sterile conditions. Following the use of mitogens to stimulate these cells to mitotically divide, colchicine is used to arrest the cells during metaphase when chromosomes are maximally visible. Giemsa banding (or G-banding) is the most commonly applied staining method to identify individual chromosomes by producing a characteristic pattern of light and dark bands on each chromosome. Abnormalities such as loss or gain of entire chromosomes, translocations of all or part of an arm of one chromosome to another or subtle changes in the banding patterns associated with various genetic disorders can be identified.
A limitation of karyotyping is that it is time-consuming, as it involves cell culture and the preparation requires several days. In addition, the resolution, which is a measure of the level of magnification of the genome, is lower than that achieved using other technologies. A standard G-banded karyotype usually has a resolution of approximately 3–5 Mb (i.e. it can detect changes of greater than three to five million base pairs). Despite these limitations, karyotyping remains the test of choice when suspecting common genetic conditions with obvious phenotypes, such as trisomy 13 (Patau syndrome), trisomy 18 (Edwards syndrome), trisomy 21 (Down syndrome) or monosomy X (Turner syndrome). Karyotyping is also the test of choice for detection of balanced structural rearrangements such as balanced translocations, and when mosaicism is suspected.
Fluorescence in situ hybridisation
Fluorescence in situ hybridisation (FISH) is a method that combines cytogenetics and molecular genetic technology.(8) Fluorescence-tagged single-stranded DNA probes are used to identify specific targeted DNA sequences during metaphase or interphase of cells. After hybridisation with the patient’s sample, the targeted regions can be visualised using a fluorescence microscope. The resolution of FISH is in the range of 100–200 kb, depending on the probe size. This renders FISH useful in the diagnosis of submicroscopic chromosomal abnormalities, including deletions, duplications and translocations. Common microdeletion syndromes that can be diagnosed using targeted FISH probes include velocardiofacial syndrome (22q11.2 deletion), Williams syndrome (7q11.23 deletion), Wolf-Hirschhorn syndrome (4p16.3 deletion), etc. FISH can be applied to interphase cells, resulting in a rapid turnaround time of 24–48 hours, allowing rapid detection of chromosomal aberrations. FISH techniques also allow the study of chromosomal aberrations in nondividing cells, for example, in cytological preparations and tissue sections. However, FISH requires prior knowledge of the specific region that might be abnormal and, therefore, has limited utility as a first-tier test for clinical diagnosis of nonspecific clinical syndromes.
Chromosomal microarray analysis
Chromosomal microarray analysis (CMA), also known as array comparative genomic hybridization, has been shown to be useful in the detection of copy number variants (CNVs), which are genomic alterations that result in an abnormal number of copies of one or more genes.(9) Compared with karyotyping, CMA has an improved resolution of approximately 50–100 kb.(10) This technology uses oligonucleotide probes capable of capturing unique genomic sequences in recurrent disease-causing CNVs associated with microdeletion and/or microduplication syndromes.(8,9) In 2010, the American College of Medical Genetics, now renamed the American College of Medical Genetics and Genomics (ACMG), recommended CMA as the first-tier clinical diagnostic test for children with unexplained developmental delay/intellectual disability, autism spectrum disorders or multiple congenital anomalies.(10) Multiple publications have reported the diagnostic yield of CMA in various cohorts of patients, with a worldwide average rate of 15%–20%, compared with that of G-banded karyotyping, with a rate of approximately 3%.(10,11) CMA has certain limitations in that it cannot identify smaller insertion-deletion (indels) or single-nucleotide variants. In addition, CMA cannot detect balanced translocations, inversions or insertions, as there is no net loss or gain of chromosomal material in these chromosome rearrangements. CMA cannot reliably detect low-level mosaicism as well.
Sanger sequencing
Sanger sequencing, which was developed by Frederick Sanger and his colleagues in the 1970s, has been the gold standard in molecular diagnostics owing to its accurate determination of sequence of nucleotide bases.(12) This method combines a DNA polymerase with a mixture of standard and chain-terminating dideoxynucleotides, which results in early termination of sequencing reactions during polymerase chain reaction (PCR).(12) The DNA fragments of varying lengths are placed into four columns on a gel and separated using gel electrophoresis, thus allowing the DNA sequence to be read. Sanger sequencing is time-consuming, as it can analyse only a single DNA segment at a time. It does not detect mosaicism below 15%–20%(13) and can miss a significant proportion of low-level mosaic variants.(14) Although Sanger sequencing is slowly being replaced by other rapid DNA sequencing technologies such as next-generation sequencing (NGS), it retains its role as an orthogonal method to confirm sequence variants identified by NGS and provide coverage for genomic regions not well-covered by NGS.
Next-generation sequencing
NGS, also known as massively parallel sequencing, is a revolutionary high-throughput sequencing technology that can simultaneously sequence multiple genes or even entire exomes or genomes in a single reaction.(15) NGS has been used in research settings for establishing the genetic basis of Mendelian diseases, especially in paediatric rare genetic disorders that can be caused by single-nucleotide variants (SNVs) or small insertions and/or deletions (indels), as NGS has an extremely high sensitivity for the detection of SNVs and indels.(16) This powerful technology has successfully identified causative genetic variants in children with suspected genetic disorders presenting as developmental delay/intellectual disability, congenital anomalies and/or autism spectrum disorders, with a yield of 25%–40% using a targeted multiple gene approach.(17,18) NGS has also been proven to be effective in identifying the genetic aetiology of children with disorders that exhibit significant locus heterogeneity, such as Noonan syndrome and related RASopathy disorders.(19) NGS is now increasingly applied in clinical paediatric practice as a diagnostic test for various genetic Mendelian disorders at different ages of presentation.
As genetic testing has become more readily available, NGS-based whole exome sequencing (WES) or whole genome sequencing (WGS) has emerged as a possible first-line diagnostic tool for paediatric rare genetic disorders with highly heterogenous phenotypes, and both methods show higher diagnostic and clinical utility than those of CMA.(5) WES examines the exons (protein-coding regions) of all 20,000 genes (approximately 1.5% of the entire genome) in which most disease-causing variants (~85%) are concentrated.(6) WGS involves the sequencing of the entire genome of three billion base pairs that include all the exons and introns (non-protein-coding regions).
WGS has the advantage of being able to detect disease-causing CNVs, structural variations, repeat expansions, and non-exonic regulatory and splicing variants.(5) The diagnostic yield of WES/WGS varies depending on the clinical features and on whether the testing approach is proband-based or trio-based (proband and both parents). For example, in a cohort of children with severe intellectual disability, the diagnostic rate for trio-based WGS was 42% and that for trio-based WES was 40%, compared with the diagnostic rate for proband-only WES, which was 28%.(15)
Table I
Summary of commonly used genetic and genomic tests.
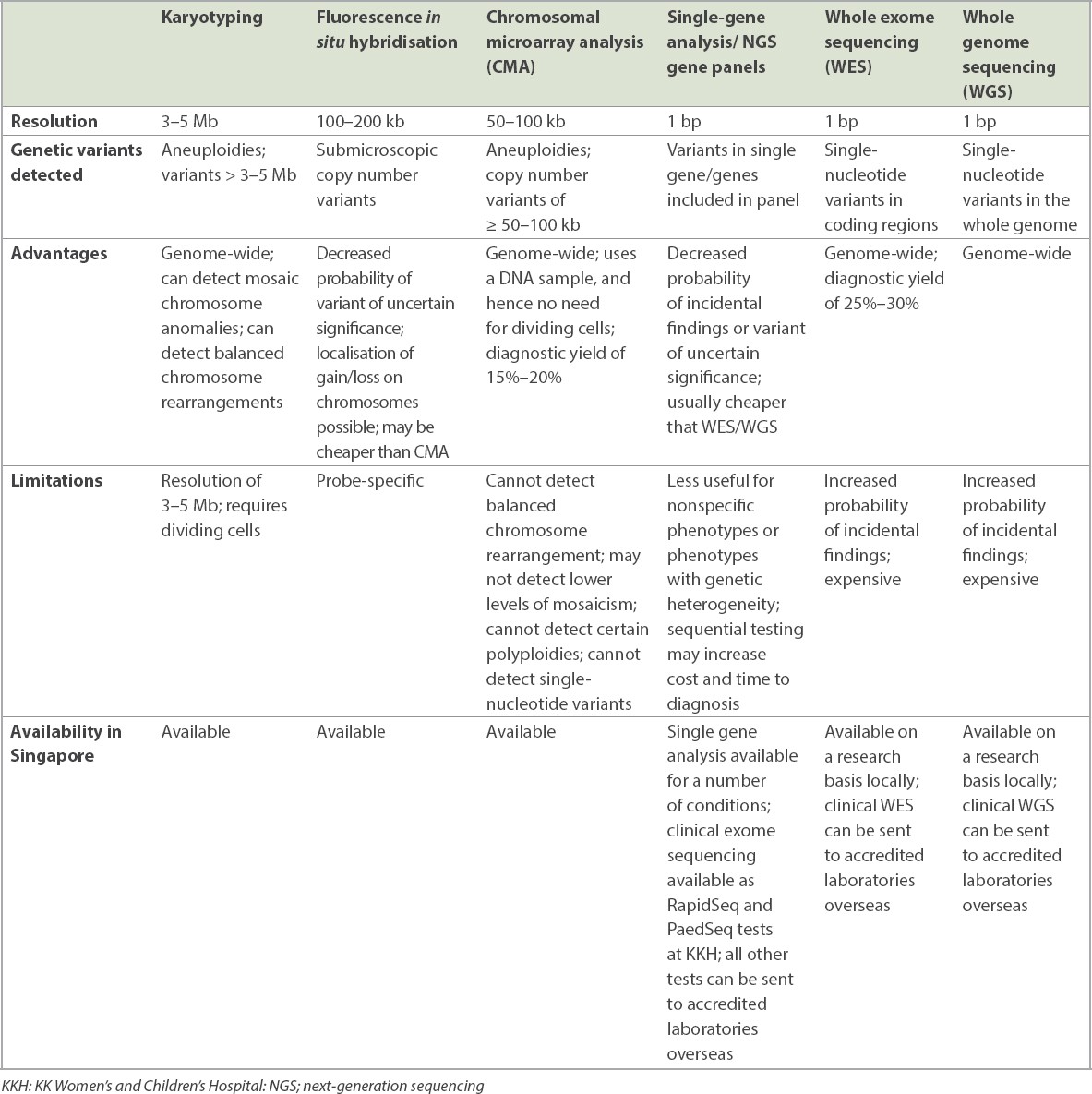
Fig. 1
(a) Karyogram shows 47,XX,+21: Female karyotype with trisomy chromosome 21 (arrow), consistent with the diagnosis of Down syndrome. (b) Fluorescence in situ hybridisation on a metaphase chromosome spread shows no signal for the test probe (HIRA) (red) and normal signal for the control probe (ARSA) (green) on one chromosome 22 homologue (solid arrow). Dotted arrow indicates the normal chromosome 22 homologue showing normal signal for both test and control probes. (c) Chromosomal microarray analysis (CMA) shows a copy number loss of 1.407 Mb at 7q11.23 at the Williams syndrome region (red bar). CMA allows delineation of the genes included in the deleted region; in this case, the deletion affects 25 genes including ELN and GTF2IRD1. (d) Integrative Genomics Viewer screenshot of next-generation sequencing reads shows a de novo variant in the KCNK9 gene (dotted box) in the proband, which is absent in the parents.
GENOMIC MEDICINE: CURRENT APPLICATION IN CLINICAL PRACTICE
Genomic technologies are increasingly being used in different clinical settings in paediatrics, including the diagnosis of rare Mendelian diseases in children using next-generation DNA sequencing.(5,6,15) Recently, the clinical utility of rapid genomic sequencing in critically ill infants and children has been reported in multiple studies.(7) Next-generation DNA sequencing also improves the diagnostic and prognostic utility of newborn screening programmes. This technological advancement has enabled sequencing of individual genes, such as those for cystic fibrosis (CFTR), to be used in newborn screening programs in many countries, including Singapore, as a confirmatory test for screen-positive infants. The emergence of novel therapeutics, including gene therapy for spinal muscular atrophy (SMA), have supported the use of PCR test for homozygous deletion of exon 7 of the SMN1 gene from dried blood samples in newborn screening programmes in several countries.(20-22)
Other emerging clinical applications of genomic sequencing include preconception carrier screening and genetic predisposition screening for disease risk assessment of adult-onset genetic conditions. Genetic carrier screening can be offered to couples to facilitate informed reproductive decision-making by identifying couples who are carriers for pathogenic variants associated with Mendelian disorders and, hence, have an increased risk of having affected offspring. Genetic predisposition testing is helpful for individuals with a positive family history or those who are at a risk of certain genetic disorders. They may be asymptomatic at the time of testing, as disease manifestation may occur later in life. Predictive testing can also identify genetic variants that increase a person’s risk of developing disorders with a genetic basis, such as genes BRCA1 and BRCA2 in breast cancer.
THE CHALLENGES
Interpretation of CNVs and sequence variants
One of the major challenges in genomic medicine is the large number of variants and their subsequent interpretation to determine the clinical significance of each variant identified in CMA and NGS-based genomic tests. Well-established guidelines from ACMG in the interpretation and reporting of constitutional CNVs and sequence variants help to standardise the interpretation of these variants across different laboratories.(23-25) Each of the variants identified is classified into various categories ranging from benign, likely benign (both are usually not reported), variant of uncertain significance (VUS), likely pathogenic and pathogenic, based on various non-overlapping criteria. The factors assessed during CNV interpretation include the genomic content, i.e. whether it contains known functionally important regions, number of genes, detailed evaluation of genomic content using published literature, public databases and/or internal lab data, and inheritance pattern.(24) In sequence variant interpretation, a variant is assessed based on the current literature describing the gene function using functional studies, computational and predictive data assessing the effect of the variant on gene function, the frequency of the variant in general population databases such as gnomAD, and the frequency of the variant in patients with clinical abnormalities using current literature and databases such as ClinVar (https://ncbi.nlm.nih.gov/clinvar/),(26) segregation and allelic data, and inheritance of the variant. The curation of each variant identified is a complex process that ensures accurate classification of the variant. However, some variants may still be classified as VUS owing to insufficient information regarding the association of these variants in genes with a genetic syndrome or phenotype. The high-throughput NGS-based genomic tests, especially WES or WGS, may potentially reveal more VUS, which may cause uncertainty among both parents of the paediatric patients and physicians regarding the appropriate clinical management to be adopted.
Multiple collaborative efforts of clinicians and scientists, including ClinGen(24,27) and ClinVar, have led to the development of various methods to improve the interpretations of these variants and consequently reduce the uncertainty.
Incidental and secondary findings
In genome-wide tests such as WES and WGS, with increasing accuracy and predictive power, there is a possibility of detecting clinically significant variants that are unrelated to the phenotype reported and clinical indication at the time of testing; these are known as incidental findings. The genetic condition incidentally identified may be an adult-onset genetic condition and may or may not have effective treatment available. The ACMG has recommended pathogenic variants in 59 genes(28) that are deemed ‘medically actionable’, known as secondary findings, to be specifically looked for and reported by the clinical laboratory if the parents of the paediatric patients wish to have the information provided. The estimated prevalence of likely pathogenic and pathogenic incidental findings in a cohort of 377 individuals in Singapore was 1.6%,(29) compared with a corresponding prevalence of 1.7% in individuals of European ancestry and 1.0%, in individuals of African ancestry.(30) These incidental or secondary findings may have implications for the patient’s future and that of his or her immediate family members. This is especially so for adult-onset conditions, which may raise concerns about negative psychosocial impacts and about depriving the child of an ‘open future’.(31) However, these incidental or secondary findings may allow at-risk parents to be identified, manage their health better and improve their ability to support their child. This information may be indirectly beneficial for the child’s welfare.(31) For example, WES performed in a child with congenital anomalies may identify an incidental finding of pathogenic variant in BRCA1, and this variant could be inherited from either parent. This means that the affected parent and other family members who are identified to carry the same pathogenic variant in BRCA1 may require surveillance and monitoring for associated cancers. Parents should be counselled carefully regarding the risk and benefits of receiving this information and should be provided the option of receiving or declining these findings.
ADOPTING GENOMIC MEDICINE IN CLINICAL PRACTICE
Personalised medicine, also known as precision medicine, is the practice of genomic medicine where an individual’s genomic data are utilised for the prevention, diagnosis and individualised treatment of his or her disease. The Singapore Undiagnosed Diseases Research Endeavour for Kids (SUREKids) study began in 2014 and was conducted at KK Women’s and Children’s Hospital (KKH) and the National University Hospital (NUH). Collaborating with the Agency for Science, Technology and Research (A*STAR) Singapore, the study aimed to explore the utility of genomic sequencing in patients with Mendelian disease.(32) Out of 196 probands whose data was reported, molecular diagnosis was made in 73 probands, with an overall diagnostic yield of 37.2%; 65 cases were diagnosed via WES (yield 37.8%) and eight, via WGS (yield 33.3%).(32) Higher diagnostic yield was observed in the cohort with global developmental delay (yield 43%), neuromuscular disorders (yield 50%) and skeletal dysplasia (yield 50%).(32) The result of this large-scale local study supports the clinical implementation of genomic sequencing in clinical practice by demonstrating high clinical utility of WES and WGS in providing accurate genetic diagnosis and management of the underlying disorder.
Clinical vignette
A six-year-old boy underwent genomic sequencing after presenting with a multisystem phenotype including poor growth, developmental delay and hepatosplenomegaly with liver transaminitis since infancy.(33) He had a history of pancytopenia when he was one month old. Despite extensive immunological and gastrointestinal investigations, no unifying diagnosis was identified. Research trio exome sequencing identified a de novo heterozygous missense variant in EIF6 (c.182G>T; p.Arg61Leu). This variant is absent in population databases, including the genomeAsia100k (https://browser.genomeasia100k.org/) and Singapore Exome Consortium, affects a highly conserved amino acid residue and is predicted to be deleterious by multiple in silico prediction software systems. However, prior to this, no disease-causative variants in EIF6 had been reported in humans. EIF6 protein is known to interact with SBDS and EFL1 protein, both of which are associated with Shwachman-Diamond syndrome (SDS). The patient’s clinical features fulfilled the diagnostic criteria of SDS according to the published international consensus guidelines,(34) and when the patient later developed frequent loose and bulky stools, pancreatic insufficiency that is associated with SDS was considered as a possible cause. This was supported by severely low stool elastase levels at <15 mcg E1/g. Subsequently, the patient was started on pancreatic enzyme replacement therapy (CREON) and since then, he has shown reduced stool frequency and improved growth. His pancytopenia improved with time, which is in contrast with the progressive bone marrow failure typically observed in patients with SDS1 due to biallelic variants in SBDS and in those with SDS2 due to biallelic variants in EFL1. Thus, our patient has a novel SDS-like phenotype with transient bone marrow failure but persistent pancreatic insufficiency.
The detailed clinical phenotyping and clinical acumen of Professor Phua Kong Boo led him to recognise the novel disease presentation in the patient. In addition, the multidisciplinary collaboration between Prof Phua, other clinical subspecialists and research collaborators allowed us to optimise the management of the patient.
ROLE OF GENOMICS EDUCATION
With the increasing use of newer genetic technologies in clinical practice, there is an urgent need to develop genomics literacy and competencies in all clinicians. Genomics education is essential for improving proficiency and skills in practical genomics, increasing confidence in identifying patients with suspected or at risk of genetic conditions for further genetic evaluation, and even ordering and interpreting genomic tests. Recognising the need for genomics education locally, an interactive workshop, Genetics Education for Healthcare Professionals, has been set up by the Genetics Service at KKH, involving faculty from KKH and NUH and supported by the SingHealth Academy and the College of Paediatrics and Child Health, Singapore. This workshop focuses on the application of a wide range of genetic and genomic tests, and practical pre- and post-test counselling skills. Its aim is to improve clinicians’ genomics knowledge including basic interpretation of genetic and genomic test results, and boost their confidence in applying genomic medicine into their clinical practice.
With the rapid progress in the field of genomics, clinicians entering medical practice now and in future will require more than a basic understanding of human genetics, which is traditionally included in the undergraduate medical curriculum in most medical schools. Enhancing exposure to genomics for medical students during both pre-clinical and clinical years of training would be important to prepare future physicians for clinical practice in the era of genomic medicine. A combination of formal and active experiential learning would be ideal to equip them with the knowledge and skills required to apply genomic medicine across a range of specialties.
FUTURE OF GENOMIC MEDICINE IN PAEDIATRICS
The Human Genome Project and DNA sequence data gathered from individuals with disorders have provided opportunities for the study of genomics and the interaction between genetic components and human diseases.(35) The current advanced genomic tests, which are more affordable, and the significant improvement in bioinformatics including variant interpretation and analysis have revolutionised clinical practice in paediatrics by providing more accurate genetic diagnoses of paediatric genetic disorders. Extensive research efforts are being made for the development of novel cell and gene therapies that could be potentially life-changing for the affected individuals. For example, Zolgensma, used for the treatment of spinal muscular atrophy (SMA), could potentially reverse the natural history of this life-limiting condition if the gene therapy is given as early as possible. This is an in vivo recombinant adeno-associated virus 9 (AAV9)-based gene therapy designed to deliver a functional copy of SMN1 gene to encode for human SMN protein, so that motor neurons can maintain their function.(36) Several clinical trials on gene therapies for different genetic conditions are ongoing, providing a new perspective in disease management, with potential curative treatment options. Genomic medicine is being more widely implemented in paediatric clinical practice; hence, it is important for clinicians to understand the basic principles of genomic medicine and familiarise themselves with the technical, ethical and legal developments in this rapidly evolving field of medicine.
CONCLUSION
Genomic testing will increasingly be integrated into mainstream paediatric practice. It is likely that genome sequencing will soon be a frontline test in most specialties, including paediatrics. Clinicians should be aware of the basic principles of genomic medicine, including its benefits and challenges. In learning to apply these advanced genomic technologies in our clinical practice, we should not neglect the importance of keeping our clinical skills sharp to phenotype our patients well and recognise which patients will benefit from genomic testing. Finally, as the story of our patient with the SDS-like condition fittingly illustrates, a collaborative spirit to work within multidisciplinary teams is of prime importance that will enable us to truly harness the power of genomic medicine to improve patient care.
ACKNOWLEDGEMENT
The authors thank Yong Min Hwee, Cytogenetics Laboratory, KKH for his expert help.