

Abstract
INTRODUCTION
Although there have been several phylogenetic studies on Plasmodium knowlesi (P. knowlesi), only cytochrome c oxidase subunit 1 (COX1) gene analysis has shown some geographical differentiation between the isolates of different countries.
METHODS
Phylogenetic analysis of locally acquired P. knowlesi infections, based on circumsporozoite, small subunit ribosomal ribonucleic acid (SSU rRNA), merozoite surface protein 1 and COX1 gene targets, was performed. The results were compared with the published sequences of regional isolates from Malaysia and Thailand.
RESULTS
Phylogenetic analysis of the circumsporozoite, SSU rRNA and merozoite surface protein 1 gene sequences for regional P. knowlesi isolates showed no obvious differentiation that could be attributed to their geographical origin. However, COX1 gene analysis showed that it was possible to differentiate between Singapore-acquired P. knowlesi infections and P. knowlesi infections from Peninsular Malaysia and Sarawak, Borneo, Malaysia.
CONCLUSION
The ability to differentiate between locally acquired P. knowlesi infections and imported P. knowlesi infections has important utility for the monitoring of P. knowlesi malaria control programmes in Singapore.
INTRODUCTION
In 2004, a cluster of malaria cases in Kapit Division, Sarawak, Borneo, Malaysia, was attributed to Plasmodium knowlesi (P. knowlesi).(1) Since this discovery of P. knowlesi infection in humans, cases have been reported in most Southeast Asian countries. In Singapore, the first reported case of P. knowlesi infection occurred in a military serviceman in 2007.(2) Isolated cases of P. knowlesi infection in Singapore have since been reported, although they are still very rare, with only two cases from the military reported between 2008 and 2013.(3) Preventive measures taken by the military, such as the use of permethrin-impregnated uniforms and advisories on the use of long-sleeved uniforms and mosquito repellents, likely contributed to this very low incidence rate. A preliminary study involving macaque sampling in various forested areas in Singapore indicated that the P. knowlesi infection was localised to the forested area in the northwestern end of Singapore; peridomestic monkeys from other parts of the island tested negative for malaria.(4) It was therefore concluded that the risk of the general population of Singapore acquiring P. knowlesi infection was small.
Phylogenetic studies on P. knowlesi using small subunit ribosomal ribonucleic acid (SSU rRNA), circumsporozoite (CSP) and merozoite surface protein 1 (MSP1)(1,5,6) showed genetic heterogeneity within P. knowlesi, but none of these genes appeared to be suitable for phylogeographic differentiation. In the study by Fong et al,(7) the authors showed phylogenetic clustering of P. knowlesi isolates based loosely on geographic region using the mitochondrial cytochrome c oxidase subunit 1 (COX1) gene as a marker. Fong et al demonstrated the ability to separate P. knowlesi isolates into three geographic clusters – Peninsular Malaysia, Sarawak and Thailand – using COX1 genetic diversity analysis.(7)
In the present study, the aforementioned four gene targets (i.e. SSU rRNA, CSP, MSP1 and COX1) were amplified from selected isolates from Singapore patients in an attempt to determine their relatedness with isolates from other parts of Southeast Asia. We also sought to determine whether there were genetic markers that were suitable for distinguishing between locally acquired and imported cases of P. knowlesi malaria. Such markers would have important utility in monitoring the effectiveness of P. knowlesi malaria control programmes.
METHODS
DNA was extracted from blood samples using the QIAamp DNA Mini Kit (QIAGEN Inc, Hilden, Germany) according to the manufacturer’s protocol for processing blood. The DNA sequences of the P. knowlesi SSU rRNA, CSP and MSP1 genes were determined using polymerase chain reaction (PCR) products that were subcloned using the pGEM®-T Easy Vector Systems (Promega, Madison, WI, USA); four recombinant plasmid clones were used for each isolate tested. Primers rPLU5 and rPLU6 were used for the amplification of a 1.3-kb fragment of the SSU rRNA gene,(1) primers PkCSP2-F and PkCSP2-R were used to amplify a 1.2-kb fragment of the CSP gene,(5) while primers PKMSP1F and PKMSP1R were used for amplification of a 1-kb fragment of the MSP1 gene.(6) A 692-bp fragment of the COX1 gene was sequenced directly from purified PCR product that was amplified using primers PKCOX1-F and PKCOX1-R.(7) Sequences were aligned using ClustalW (EMBL-EBI, Hinston, Cambridgeshire, UK) and Lasergene MegAlign (DNASTAR, Madison, WI, USA). Phylogenetic trees were constructed using the maximum parsimony method with 1,000 bootstrap replicates, as implemented by the MEGA software version 6.0.(8) The CSP, MSP1, SSU rRNA and COX1 gene sequences for the non-Singapore isolates were obtained from GenBank.
RESULTS
The CSP, SSU rRNA and MSP1 phylogenetic analysis showed no obvious difference between the P. knowlesi isolates from Singapore and those from other Southeast Asian countries (Figs.
Fig. 1
Phylogenetic diagrams show the genetic diversity of (a) the CSP gene sequences from the P. knowlesi-positive samples from Singapore and Malaysia; (b) the SSU rRNA gene sequences from the P. knowlesi-positive samples from Singapore, Thailand and Malaysia; and (c) the MSP1 gene sequences from the P. knowlesi-positive samples from Singapore, Thailand and Malaysia. No distinct clustering in the isolates from Singapore (bold text) was observed for all three genes. The phylogenetic tree was constructed using the maximum parsimony method. On the branches are bootstrap percentages based on 1,000 replicates. The scale bar at the base of the figure depicts the number of nucleotide differences. The phylogenetic analysis was conducted using MEGA6.(8)
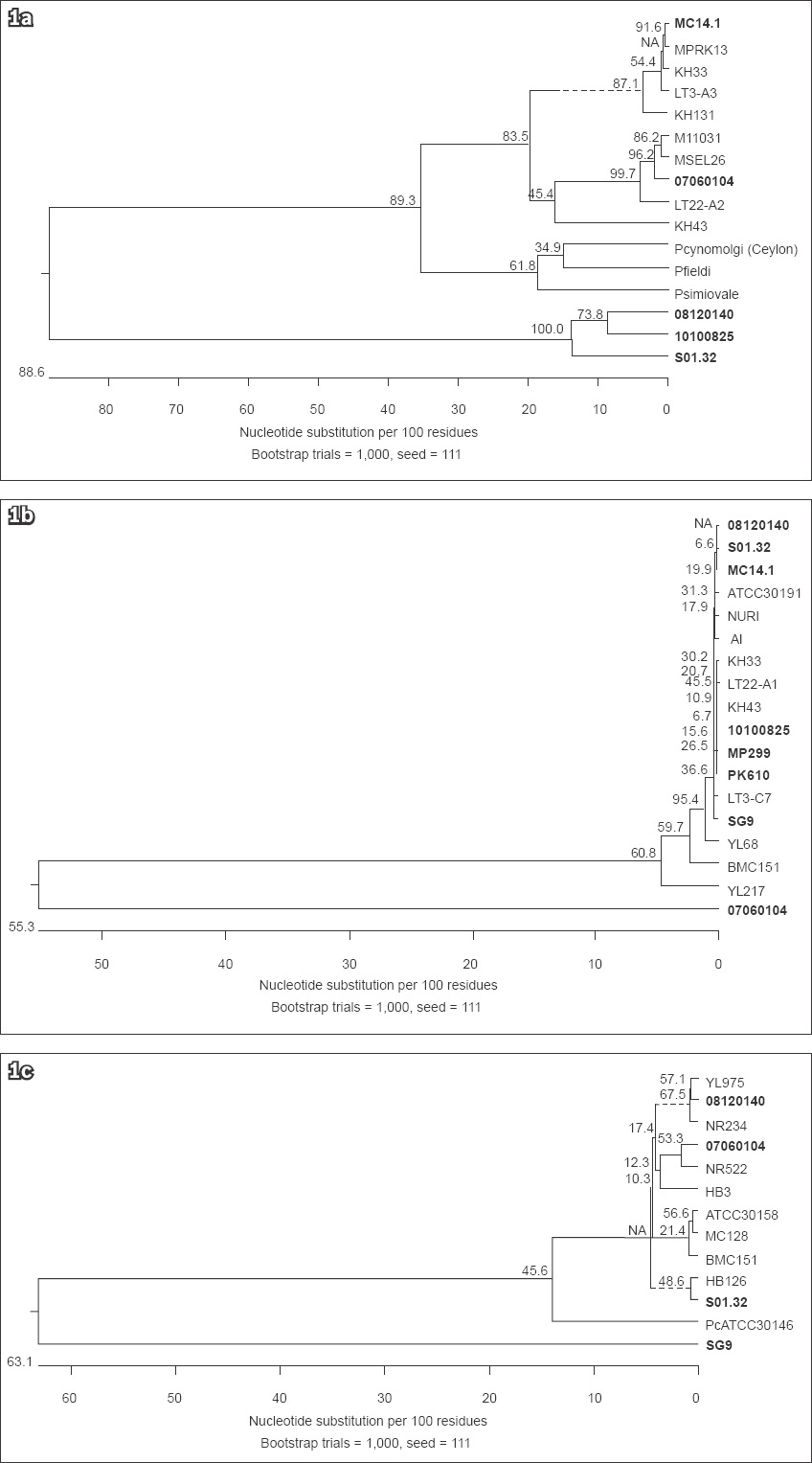
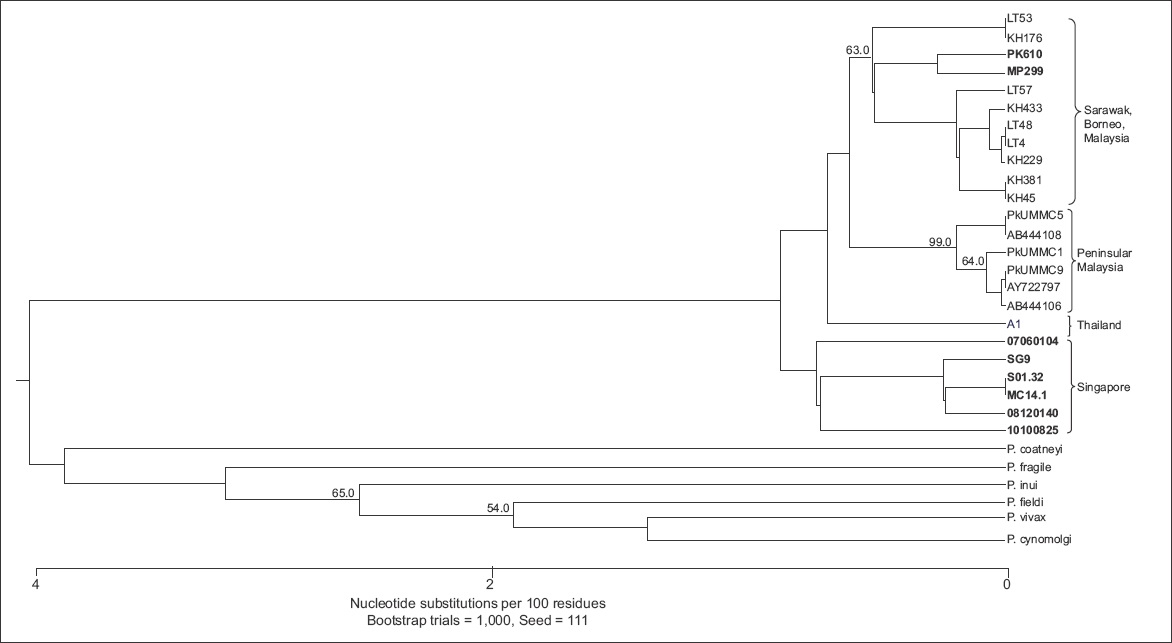
However, the COX1 phylogenetic analysis showed some differences between the majority of the P. knowlesi isolates from Singapore, and the isolates from Malaysia and Thailand. The majority of the P. knowlesi isolates from Singapore formed a separate clade (
Fig. 2
Phylogenetic diagram shows the genetic diversity of the COX1 gene sequences from the P. knowlesi-positive samples from Singapore, Thailand and Malaysia. A distinct cluster was observed in the isolates from Singapore (bold text). The phylogenetic tree was constructed using the maximum parsimony method. On the branches are bootstrap percentages based on 1,000 replicates (only those above 50% are shown). The scale bar at the base of the figure depicts the number of nucleotide differences. The phylogenetic analysis was conducted using MEGA6.(8)
DISCUSSION
Sequencing and phylogenetic analysis of the COX1 gene from selected P. knowlesi isolates from Singapore showed that these isolates formed a distinct clade, separate from the clades formed by the P. knowlesi isolates from Thailand, Peninsular Malaysia and Sarawak, although it was closer to the clade formed by the isolates from Thailand. Our COX1 phylogenetic analysis also showed that two of the P. knowlesi isolates from Singapore (PK610 and MP299) clustered with the isolates from Sarawak, indicating that they were likely to be imported cases of P. knowlesi infection from Malaysia. This is possible as both cases originated from the general public and the patients presented with malaria in public hospitals. Unfortunately, the case and travel histories of isolates MP299 and PK610 were not available for further analysis and confirmation.
Through the use of COX1 gene analysis, it may be possible to differentiate between local and imported P. knowlesi infections. This is of importance to local health authorities who carry out P. knowlesi malaria control activities. The similarities between the isolates from Singapore and Thailand also need to be investigated further, as the two countries are separated by Peninsular Malaysia, which is more than 700 km in length. The import of P. knowlesi from Singapore to Thailand, or vice versa, in the distant past is therefore a possibility. Research is currently in progress to develop single nucleotide polymorphism PCR assays that target the 295 and 646 allele for the COX1 gene, so as to enable faster differentiation of local and imported P. knowlesi infections prior to sequencing for confirmation.
In conclusion, the present study has shown that although it would be difficult to distinguish between locally acquired and imported P. knowlesi malaria using the genetic information of SSU rRNA, MSP1 and CSP, it may be possible to do so using the COX1 gene.
ACKNOWLEDGEMENTS
This study was funded by the Ministry of Defence, Singapore, and DSO National Laboratories, Singapore.