

Dear Sir,
Bland soft tissue spindle cell neoplasms are frequently encountered by pathologists in their daily work. Perineurioma of the soft tissue is a benign peripheral nerve sheath tumour composed of bland spindle cells with storiform and whorls within a collagenous to myxoid matrix.(1)
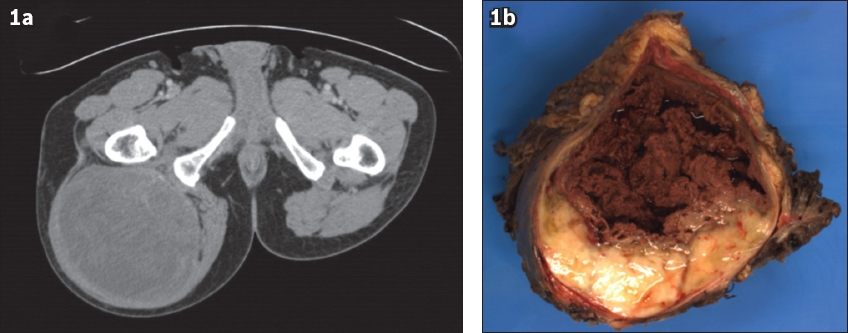
Herein, we describe the case of a 53-year-old man who was admitted to our hospital for a right gluteal mass. Computed tomography (CT) revealed a well-circumscribed heterogeneously enhancing tumour occupying the right gluteal region – which was possibly intramuscular in location – with the right sciatic nerve not well-delineated from the mass (
Fig. 1
(a) CT image shows a well-circumscribed heterogeneously enhancing tumour occupying the right gluteal region, which is possibly intramuscular in location. (b) Photograph shows the gross appearance of the surgical specimen, which demonstrates a solid cystic appearance with myxoid cut surface.
Microscopic examination revealed a well-circumscribed, unencapsulated nodular lesion composed of a proliferation of bland to bipolar spindle cells in fascicles within a collagenised and myxoid oedematous stroma. Focal myxoid change and whorled growth pattern were identified. No nuclear atypia or increased mitoses suggestive of malignancy was seen. The spindle cells were negative for many epithelial and mesenchymal markers, such as AE1/3, MNF116, SMA, desmin, h-caldesmon, CD34, S100, Glut-1, MDM2 and CDK4. Only epithelial membrane antigen (EMA), a nonspecific epithelial marker, was tested positive (
Fig. 2
Photomicrographs show (a) proliferation of bland to bipolar spindle cells in fascicles with whorled growth pattern and (b) focal myxoid change (Haematoxylin & eosin, × 20). These cells are positive for (c) epithelial membrane antigen and (d) MUC1.
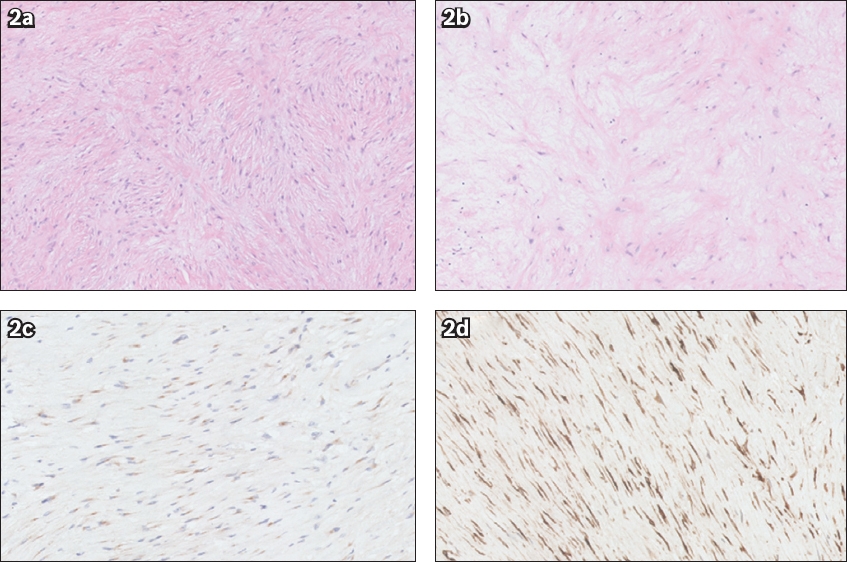
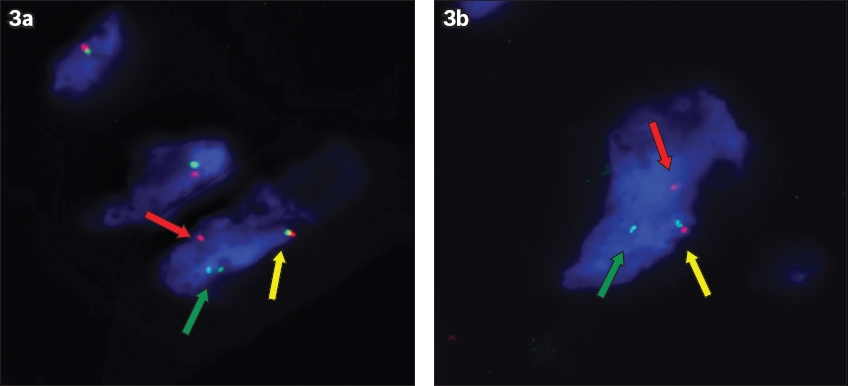
However, on subsequent review by a sarcoma pathologist, the lesional cells were found to be positive for MUC4 immunohistochemical staining, which is typically not seen in perineuriomas.(2) FUS gene rearrangement was detected using fluorescence in situ hybridisation (FISH) (
Fig. 3
(a & b) Images show interphase nuclei of the tumour cell demonstrating rearrangement of the FUS gene using fluorescence in-situ hybridisation. The FUS dual color break-apart probe consists of two direct-labelled probes hybridised to 16p11.2 band. The orange fluorochrome direct-labelled probe hybridised proximal to the FUS gene while the green fluorochrome direct-labelled probe hybridised to the distal end of the FUS gene. The signal pattern in these images consist of one fusion (yellow arrow), one orange (red arrow) and one green (green arrow) signals, indicating disruption of the FUS gene at 16p11.2.
LGFMS was first described by Evans in 1987(3) as a histological deceptively bland fibromyxoid neoplasm with metastatic potential and a propensity for late local recurrence.(1,3,4) As demonstrated in this case, LGFMS may be difficult to identify histologically. Most often, the cases are misdiagnosed as benign conditions with bland spindle cells and focal myxoid change such as perineurioma, neurofibroma, nodular fasciitis and many other entities.(3-5) This can occur especially in centres where general pathologists with no subspecialisation see most of the soft tissue cases, or where there is a lack of sarcoma pathologists, multidisciplinary discussion, and access to appropriate immunohistochemical stains or molecular tests. Immunohistochemistry plays a vital role in differentiating this tumour from other entities. MUC4, a transmembrane glycoprotein that functions in cell signalling pathways, is aberrantly expressed in LGFMS, with high sensitivity and specificity.(2) EMA has also been found to be positive in 80% of LGFMS.(1)
The cytogenetic hallmark of LGFMS is the t(7;16)(q33;p11), resulting in FUS-CREB3L2, which is present in 81%–96% of cases.(6,7) A rare variant has shown evidence of t(11;16)(p11;p11), resulting in a FUS-CREB3L1 fusion.(7) In a study by Mertens et al,(7) 22 out of 23 (96%) low-grade fibromyxoid sarcoma cases demonstrated a FUS-CREB3L2 fusion that was not seen in the other fibrous and myxoid neoplasms tested, thus supporting the sensitivity and specificity of FUS translocation. However, FUS translocation can also be seen in myxoid liposarcoma (FUS-DDIT3), which microscopically shows paucicellular spindle cells in a myxoid stroma with chicken-wire vasculature and signet-ring lipoblasts; angiomatoid fibrous histiocytoma (FUS-ATF1) demonstrating highly cellular sheets of histiocytoid cells with haemorrhagic cyst-like spaces surrounded by a dense peripheral cuff of inflammatory cells; and rarely, in Ewing sarcoma (FUS-ERG) with a highly malignant small-round-cell appearance.(1) Rearrangements of the FUS gene may be detected using FISH or reverse transcription polymerase chain reaction (RT-PCR),(6) which facilitates confirmation of unusual histological variants of LGFMS. RT-PCR shows high specificity in determining the presence of certain fusion transcripts, but is not able to identify those with different or novel fusion partners. For example, an FUS-CREB3L2 RT-PCR would not be able to identify those LGFMS cases with the rare FUS-CREB3L1 fusion. This is in contrast with the FUS FISH, which demonstrates high sensitivity in detecting neoplasms with FUS translocations.(6) Hence, although FUS FISH is excellent for identifying a low-grade fibromyxoid sarcoma, the caveat is that other neoplasms with FUS translocation, such as those discussed above, must be excluded.
In conclusion, due to its variable and bland morphology, LGFMS remains a diagnostic challenge and a potential pitfall for a wide range of low-grade fibrous and myxoid lesions (including fibromatosis, fasciitis, myxoma and perineurioma), especially in cases with limited tissue such as in core biopsies. However, having a lower threshold to implement immunohistochemistry and genetic analysis to differentiate LGFMS from other entities, especially if demonstrating bland spindle cells and myxoid changes histologically, will aid in ensuring that a correct diagnosis is reached. This would then prompt further assessment, which includes ensuring adequate surgical excision with margin clearance, a full oncological assessment to detect the presence of metastasis and adjuvant therapy if needed, as well as a longer follow-up due to the propensity for late local recurrence and the metastatic potential of the disease.
Yours sincerely,