

His mother took three-year-old Brian Tan to your clinic because she had a lot of difficulty waking him up that morning. He was awake but irritable, and she noted that he was refusing his breakfast.
WHAT IS ENCEPHALOPATHY?
Encephalopathy is a clinical state characterised by an alteration of consciousness, behaviour and/or cognition. It can present with lethargy and drowsiness, or conversely with a heightened state of agitation and confusion. It arises from diffuse brain dysfunction that can result from myriad underlying conditions, ranging from primary neurologic to systemic disorders. While encephalopathy more commonly stems from a global brain insult, it can present with a range of focal neurological manifestations such as seizures, visual disturbances, speech abnormalities, motor weakness, and sensory and autonomic deficits. Encephalitis refers specifically to brain inflammation, usually associated with fever, with evidence of cerebrospinal inflammation (pleocytosis and raised protein levels) and/or neuroimaging findings of encephalitis.
HOW RELEVANT IS THIS TO MY PRACTICE?
Altered behaviour is common in children and has many causes. In the general clinic setting, it is important to pick up common clinical mimics of encephalopathy that can be managed without immediate referral to the hospital. These include post-ictal drowsiness from simple febrile seizures, and metabolic derangements such as hypoglycaemia. Conversely, it is also important to identify red flags in the history and physical examination that necessitate either activation of emergency services or an urgent referral to the neurologist for further workup.
In particular, encephalitis accounts for a significant proportion of encephalopathy cases that persist beyond 24 hours, and can result in significant morbidity and mortality. The worldwide incidence of encephalitis ranges from 3.3 to 10.7 per 100,000 children per year.(1-3) Arriving at an accurate diagnosis for the underlying cause is critical because many causes are reversible, and early specific treatment confers a better prognosis.
WHAT CAN I DO IN MY PRACTICE?
In this primary care approach to encephalopathy in paediatric patients, we propose looking at the main differential diagnoses by age group and time course of onset. A comprehensive history is vital, especially from the caregiver, as patient accounts may be limited due to their age and clinical status. Key features in the physical examination should be specifically looked out for. These provide a framework to guide investigations and management (
Table I
Recommended approach to history and physical examination.
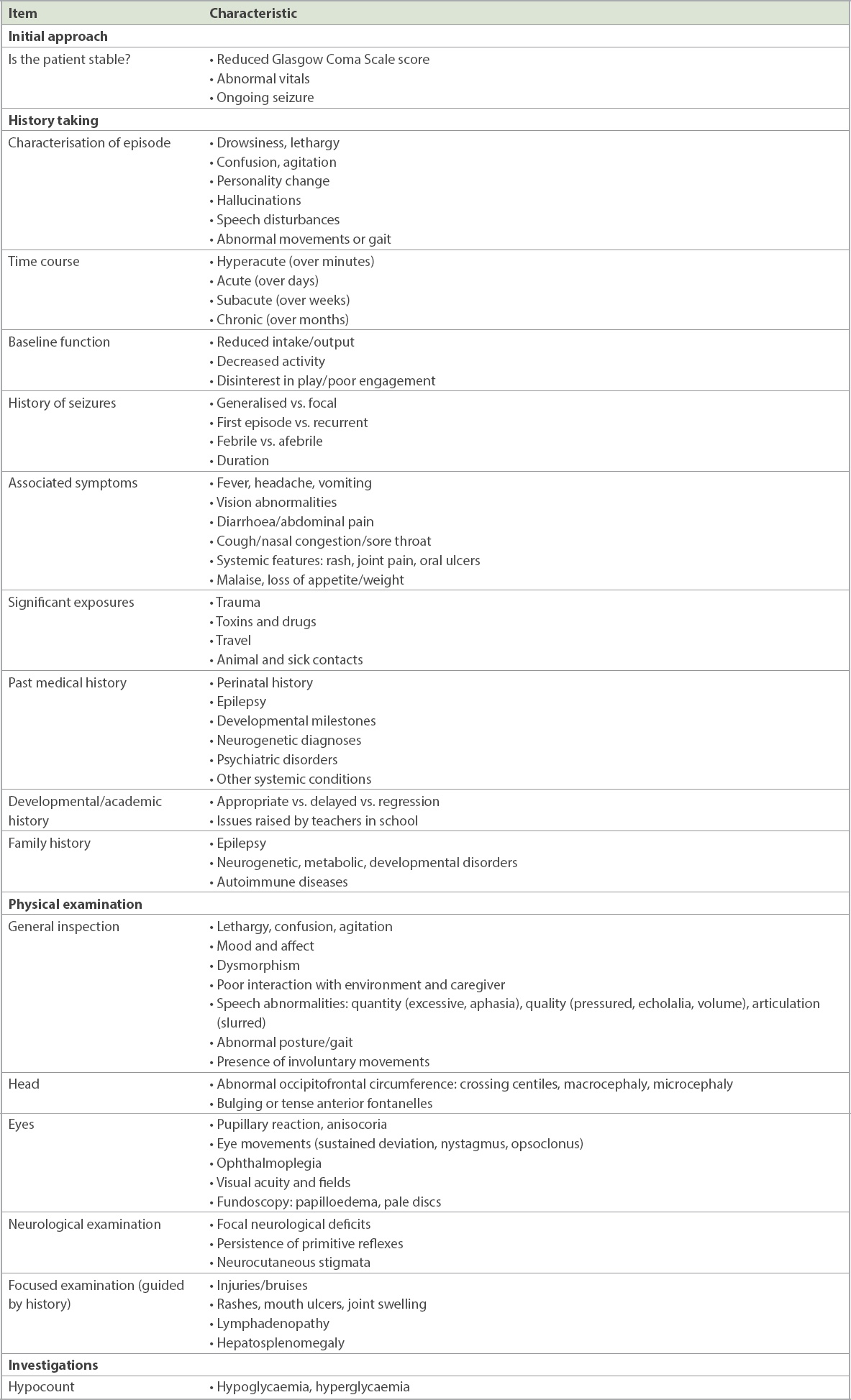
Pertinent features in history taking
Careful attention needs to be paid to the onset, duration and progression of symptoms (
Table II
Primary neurological conditions causing encephalopathy.
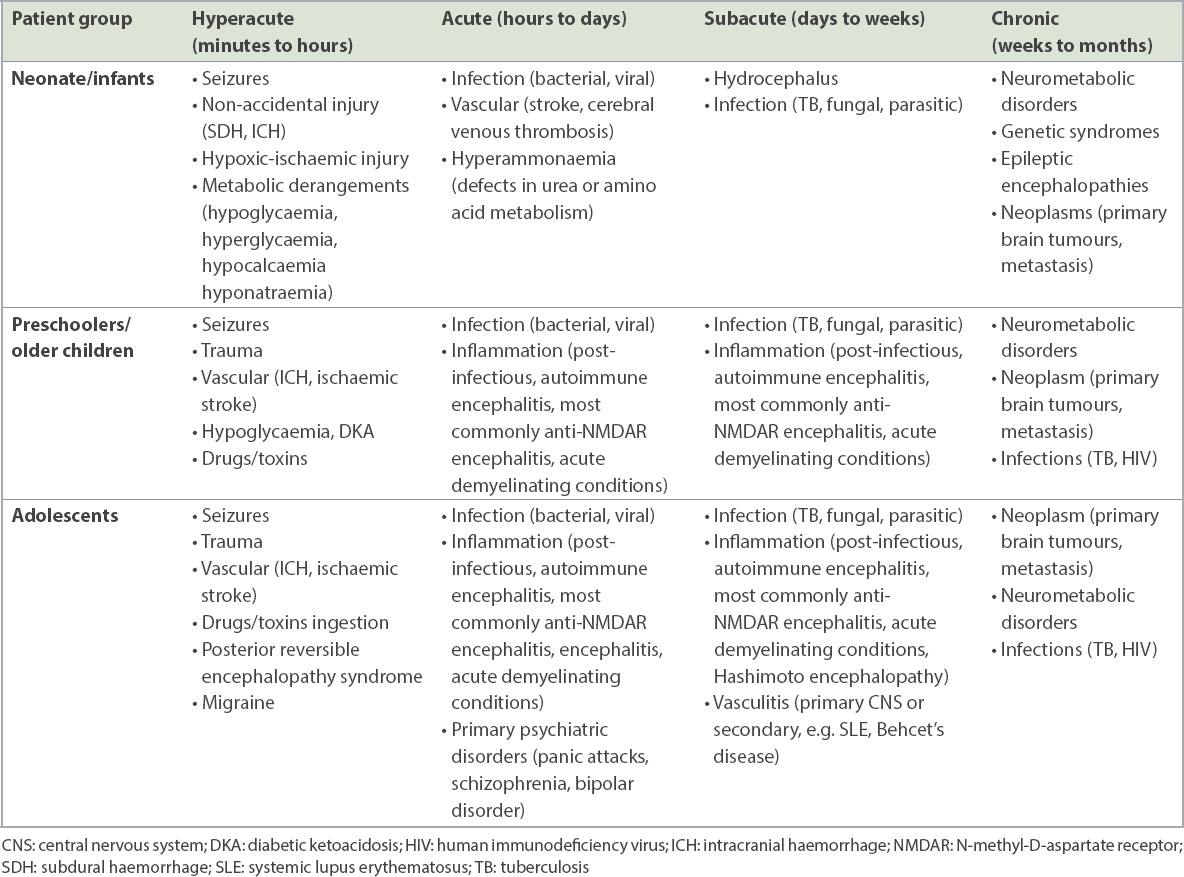
In children, functionality and deviation from baseline behaviour is critical in ascertaining the presence, and degree, of encephalopathy. It is thus important to elicit this information from the main caregiver specifically, if possible; this may not necessarily be the parent who brought the child but a domestic helper or grandparent. The caregiver’s reports of irritability, lethargy or drowsiness should always be taken seriously, especially if these symptoms are disproportionate to any fever spikes. In younger patients, aversion to feeds, disinterest in toys and play, and decreased activity may be the only manifestations of encephalopathy. Infants with poor intake (usually considered less than 50% of normal) and dehydration (increased thirst, reduced urinary output) due to an otherwise simple viral illness are nevertheless predisposed to hypoglycaemia or cerebral venous thrombosis, both of which can also result in altered mentation. Even if the child appears well during the point of clinical contact, there should be a high index of suspicion for delirium, which presents with fluctuating levels of alertness. It may be prudent in these cases to advise admission for monitoring.
Seizures are sometimes the first presentation of encephalopathy but are far more commonly caused by simple febrile seizures or epilepsy. As a general rule, a simple febrile fit in a child older than 18 months, who has no red flags (i.e. normal physical/neurological examination, duration < 15 minutes, no recurrence within 24 hours, no focal lateralising signs, normal developmental milestones, no prior head trauma/central nervous system infections) and has returned to baseline function, can be managed in the primary care setting; all other patients should be referred to the emergency department. It should also be noted that neonatal seizures can be subtle and are usually not generalised in view of immature cerebral motor pathways.(4) However, if episodes are recurrent and stereotypical, clinicians need to have a high index of suspicion for prompt referral. Caregivers should be encouraged to take videos of suspected episodes, as reviewing them is helpful in characterising the semiology and directing further investigation.
The presence of fever suggests an infective or infection-associated encephalitis, which is often preceded or accompanied by respiratory or gastrointestinal symptoms. Correspondingly, the commonest pathogens detected with encephalitis are Streptococcus, influenza virus, Mycoplasma pneumoniae, Escherichia coli, enterovirus and rotavirus.(5) Travel and contact history (including animals) is important in screening for more atypical infections. In older teens, systemic features (photosensitive rash, joint pain, ulcers) should be sought, as autoimmune diseases such as systemic lupus erythematous tend to present in this age group.
In previously healthy children, an acute to subacute onset of neuropsychiatric symptoms (personality change, auditory hallucinations) accompanied by a suggestive clinical phenotype that includes movement abnormalities, speech impairment, seizures, mood lability and sleep dysregulation raises the suspicion of autoimmune encephalitis.(6,7) In the last decade, autoimmune encephalitis has been shown to rival the frequency of viral aetiologies(8) and should be considered early in the presentation of encephalopathy. Many are seronegative, but the most common autoantibody in children targets the N-methyl-D-aspartate receptor (NMDAR). A timely diagnosis will avoid unnecessary diagnostic and treatment costs and permit earlier directed therapy.(9)
Finally, environmental risk factors should be explored. In the younger child, caregivers should be questioned on the possibility of accidental toxin exposure (e.g. child left unattended or medications/toxins within reach). In the adolescent, the possibility of alcohol use, drug use or intentional overdose should not be overlooked.
Clinical examination
The first priority of the clinical examination is to ensure the child is haemodynamically and neurologically stable, with a normal, age-appropriate heart rate, respiratory rate, oxygen saturations and blood pressure (
Table III
Normal vital parameters by age.
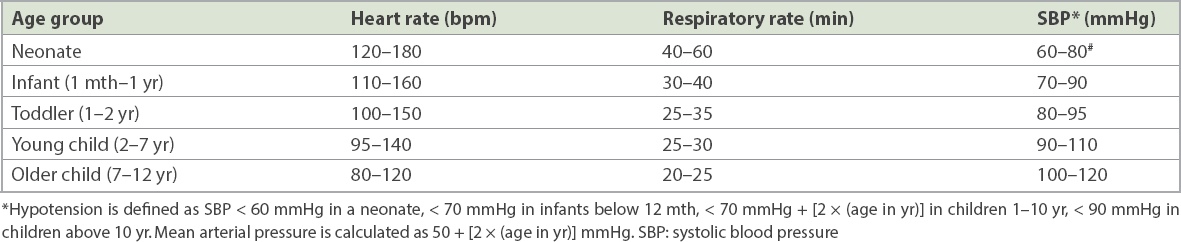
Table IV
Glasgow Coma Scale (GCS).
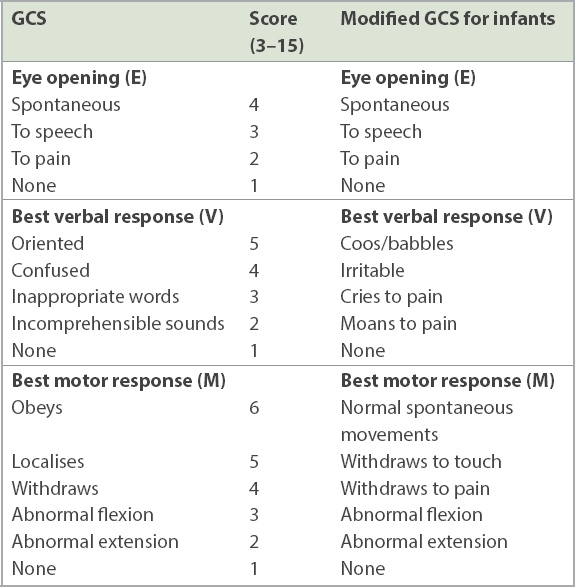
It is important to note that while the GCS score is helpful, it should not be the only measure of an altered mental state. Encephalopathy can be present even with a GCS score of 15, as the score does not take into consideration other behavioural or cognitive changes such as mood lability and higher-level executive functioning. The inclusion of a neurocognitive test such as the Mini Mental State Examination for the older paediatric patient can reveal subtle cognitive dysfunction that would be missed at first impression (
Box 1
Mini Mental State Examination:

As the clinical history is being obtained from the caregiver, an observant clinician can simultaneously glean valuable information from watching the patient in the caregiver’s lap or at play. This is especially helpful in younger children who may be fretful and refuse more direct examination. The child’s resting posture, movement and interaction should be observed, noting poor tone, asymmetry of limb movement, abnormal posturing, lack of interaction or poor eye contact with caregivers. In an older patient, gait and movement abnormalities should be specifically looked for, even before performing a formal neurological examination. Choreoathetoid and other dyskinetic movements of the face, mouth, tongue and limbs and/or catatonic features (staring, minimal responsiveness or excessive hyperactivity, rigid posturing) are typical presentations for autoimmune encephalitis if drug ingestion has been ruled out.(6,7)
A detailed examination of the eyes and head can reveal important clues. Small or large pupils that are sluggish to light in the absence of a dropping GCS score may suggest drug ingestion. Persistent gaze deviation may be a sign of ongoing seizure. An isolated sixth nerve palsy and/or papilloedema points to raised intracranial pressure, while ophthalmoplegia should prompt careful examination of the other cranial nerves for better localisation of the pathology. Reduced visual acuity with red desaturation is highly suggestive of optic neuritis in the setting of a demyelinating disorder, such as neuromyelitis optica or multiple sclerosis.
Occipitofrontal circumference (OFC) should be routinely measured in all children aged two years and under. Serial OFCs that rapidly increase and cross centiles raise the suspicion of hydrocephalus or a space-occupying lesion, while microcephaly (OFC < third centile) or macrocephaly (OFC > 97th centile) could be clues of underlying perinatal insults or neurogenetic disorders. A tense or bulging anterior fontanelle in a non-crying infant is also indicative of raised intracranial pressure.
A developmental assessment together with the full neurological examination should be performed in infants and younger children, especially those with more chronic symptoms, as this may be a manifestation of underlying malignancy or neurometabolic disorder. Developmental regression, defined as a plateauing or loss of milestones already acquired, is a red flag for early specialist referral.
Investigations
In the primary care setting, a quick hypocount would exclude any reversible glucose derangement that can account for the altered mental status. Hypoglycaemia is generally defined as serum glucose less than 3.5 mmol/L. It is a particularly important diagnosis to exclude in neonates and infants, as delayed treatment can result in neuronal injury in a vulnerable young brain. Hypoglycaemia can also be the first manifestation of an underlying metabolic or endocrine disorder. Hyperglycaemia in the setting of acidosis is suspicious for diabetic ketoacidosis (serum glucose > 11 mmol/L, pH < 7.3, HCO3 < 15 mmol/L), which can present with lethargy, drowsiness and Kussmaul’s breathing.
Management
Stable patients (i.e. GCS 15 and normal vitals with no focal neurological deficits) with more chronic behavioural change or developmental delay/regression may be referred for an early neurology consult. Otherwise, most patients need to be referred to an emergency department.
While there may be limited resuscitative ability in the general clinic, some simple measures can be instituted while awaiting transfer. Hypoglycaemia can be easily treated with oral feeds (simple sugars, e.g. sweets, juice or a glucose gel) or an intravenous 2 mL/kg bolus of 10% dextrose if the child is symptomatic or unable to tolerate orally. There is often concomitant dehydration in diabetic ketoacidosis, but fluid boluses should be avoided unless there is haemodynamic shock, as rapid rehydration increases the risk of cerebral oedema. Ongoing seizure lasting more than five minutes can be aborted with either rectal diazepam 0.4 mg/kg (max dose 10 mg) or intravenous lorazepam 0.1 mg/kg (max dose 4 mg). Each dose can be repeated once with a maximum of two doses of benzodiazepines using either route. The child should be placed in a lateral position on a level surface, preferably in a well-ventilated area, and oxygen can be administered to keep saturations above 95%. These interim measures help to stabilise the patient before the transfer.
Further management in the tertiary setting
Once the child has been referred and stabilised appropriately, more detailed investigations and subsequent management can be undertaken in the tertiary setting. Diagnostic studies in hospitals are generally approached in two stages. A first-line screen includes basic haematology, biochemistry and pathogen identification in blood, cerebrospinal fluid and other specific sites dictated by the initial assessment. Autoimmune and metabolic screening is added if there was developmental delay, systemic symptoms or a significant family history at presentation. If there are persistent symptoms or clinical deterioration, investigations are extended to include a wider search for atypical organisms, such as TB, and more specialised tests for central nervous system inflammation, such as anti-NMDAR receptor antibodies.
Computed tomography of the brain is quick and detects intracranial bleeds and focal lesions, but magnetic resonance imaging is the preferred imaging modality for accurate and sensitive identification and characterisation of various pathology with no risk of ionising radiation. Demyelinating disorders and an increasing number of viral-associated encephalitis now have recognisable clinico-radiological phenotypes, which facilitates early diagnosis and treatment.(10,11) Magnetic resonance spectroscopy and magnetic resonance angiography are also helpful in neurometabolic and vascular aetiologies.(7) Electroencephalography can be utilised to detect subclinical seizures, assess the degree of encephalopathy and localise areas of pathology. Genetic testing is also increasingly employed to aid in diagnosis, especially in epileptic encephalopathies and neurometabolic disorders.
Specific treatment will subsequently be guided by the likely aetiology. In acute-onset encephalopathy, early broad-spectrum antimicrobials may be initiated while awaiting diagnostic studies. First-line therapy consists of intravenous ceftriaxone and acyclovir, with the addition of ciprofloxacin if mycoplasma infection is suspected. Immunotherapy with intravenous methylprednisolone and/or immunoglobulin is given for probable autoimmune encephalitis. General neuroprotective measures are instituted to prevent secondary cerebral injury. More chronic encephalopathy will require detailed workup and neurorehabilitative intervention to optimise the patient’s functional status.
TAKE HOME MESSAGES
-
Encephalopathy is a broad term characterised by impaired awareness, cognition and behaviour, and can be caused by a wide range of conditions from systemic to primary neurologic disorders.
-
Common clinical mimics of encephalopathy include post-ictal drowsiness, hypoglycaemia and toxin ingestion. A good history and physical examination should be able to identify these causes.
-
A useful approach to narrowing the differential diagnoses is determining the onset and evolution of symptoms in specific age groups, followed by thorough and directed history taking and examination that includes GCS scoring and the Mini Mental State Examination.
-
As neonatal encephalopathy and seizures can present subtly, a high index of suspicion is required. Video-recording of episodes can be very useful in characterising their semiology.
-
A simple hypocount or point-of-care test can be performed in the primary care clinic setting to rule out hypoglycaemia and diabetic ketoacidosis.
-
Acute seizure management involves placing the patient in a lateral position with supplemental oxygen in a well-ventilated room. If the seizure persists beyond five minutes, rectal diazepam or intravenous lorazepam should be administered.
You called Brian’s main caregiver, his grandmother, and found out that his temperature had been spiking since the day before. He was also taking less than half his usual share of food, and was lethargic and minimally interactive for most of the day, even during the times when he was afebrile. In view of the above, you quickly sent Brian via ambulance to the children’s emergency department after ensuring that he was otherwise stable. Two months later, his grandmother thanked you for your prompt assessment and transfer. It meant that Brian received timely treatment for his viral encephalitis and subsequently had a full recovery. He suffered no long-term complications and was back to his usual cheerful self.
SMJ-61-632.pdf