

Abstract
A 68-year-old man presented with a three-week history of rapidly progressive dementia, gait ataxia and myoclonus. Subsequent electroencephalography showed periodic sharp wave complexes, and cerebrospinal fluid assay revealed the presence of a 14-3-3 protein. A probable diagnosis of sporadic Creutzfeldt-Jakob disease was made, which was further supported by magnetic resonance (MR) imaging of the brain showing asymmetric signal abnormality in the cerebral cortices and basal ganglia. The aetiology, clinical features, diagnostic criteria, various MR imaging patterns and radiologic differential diagnosis of sporadic Creutzfeldt-Jakob disease are discussed in this article.
CASE PRESENTATION
A 68-year-old Caucasian man with no significant past medical history presented to the emergency department with a three-week history of progressive memory loss, involuntary muscular twitches deemed to represent myoclonus, and an unsteady gait. Electroencephalography (EEG) revealed periodic sharp wave complexes and cerebrospinal fluid (CSF) analysis revealed the presence of a 14-3-3 protein. What do the magnetic resonance (MR) images (Figs.
Fig. 1
(a & b) Fluid-attenuated inversion recovery (FLAIR) and (c & d) diffusion-weighted images.
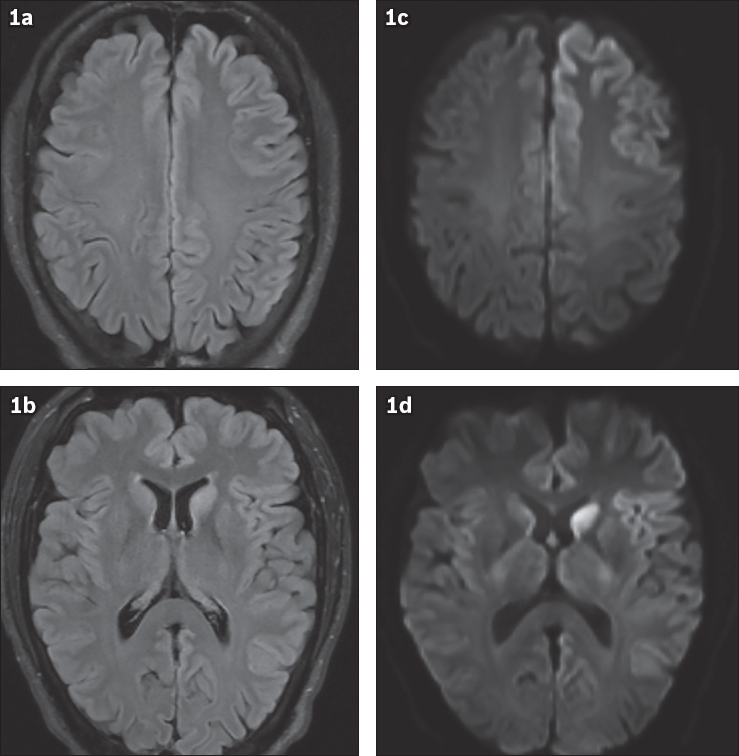
IMAGE INTERPRETATION
Axial fluid-attenuated inversion recovery (FLAIR) images show hyperintense signal in the left caudate nucleus and asymmetric signal abnormality in the cerebral cortices, worse on the left side, primarily affecting the left frontal lobe, left insula and left temporal-parietal lobes (Figs.
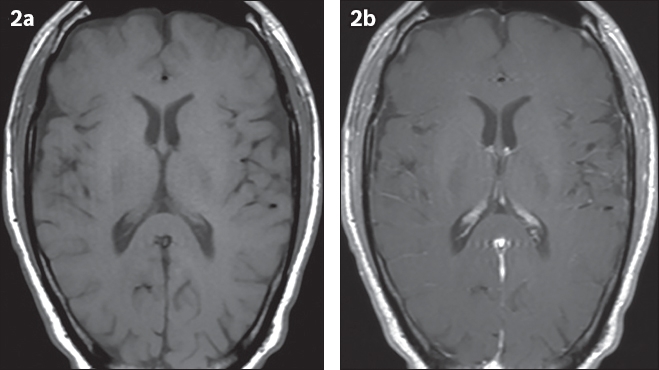
Fig. 2
(a) Pre- and (b) post-gadolinium T1-weighted images.
DIAGNOSIS
Sporadic Creutzfeldt-Jakob disease (sCJD).
CLINICAL COURSE
The patient’s clinical condition progressively deteriorated over a period of 18 months; he eventually became unconscious, requiring intubation with mechanical ventilation. The course was complicated by sepsis and the patient eventually died. While no autopsy was performed, the clinical and paraclinical tests were sufficient to make a probable diagnosis of sCJD.
DISCUSSION
Creutzfeldt-Jakob disease (CJD) is a group of human prion diseases alternatively known as transmissible spongiform encephalopathies. It was first described independently by Alfons Maria Jakob and Hans Gerhard Creutzfeldt in 1920. The mechanism of prion diseases was formally proposed by Stanley Prusiner in 1982, which eventually earned him the 1997 Nobel Prize in Medicine. It is now understood that human prion diseases are caused by an alteration of the naturally existing, detergent-soluble and protease-sensitive prion protein into an abnormally folded, detergent-insoluble and protease-resistant protein, termed scrapie prion protein (PrPSc), in the brain.(1) The process propagates through self-replication of the newly formed PrPSc that eventually results in spongiform degeneration and neuronal loss. sCJD is by far the most common and best understood subtype, accounting for about 85% of overall CJD cases, with the remaining consisting of familial (10%–15%) and acquired (1%) subtypes, such as fatal familial insomnia and variant CJD, respectively.(2) sCJD has an incidence of about one case per million people per year worldwide and peaks between the ages of 65 and 74. No gender predilection exists.(3)
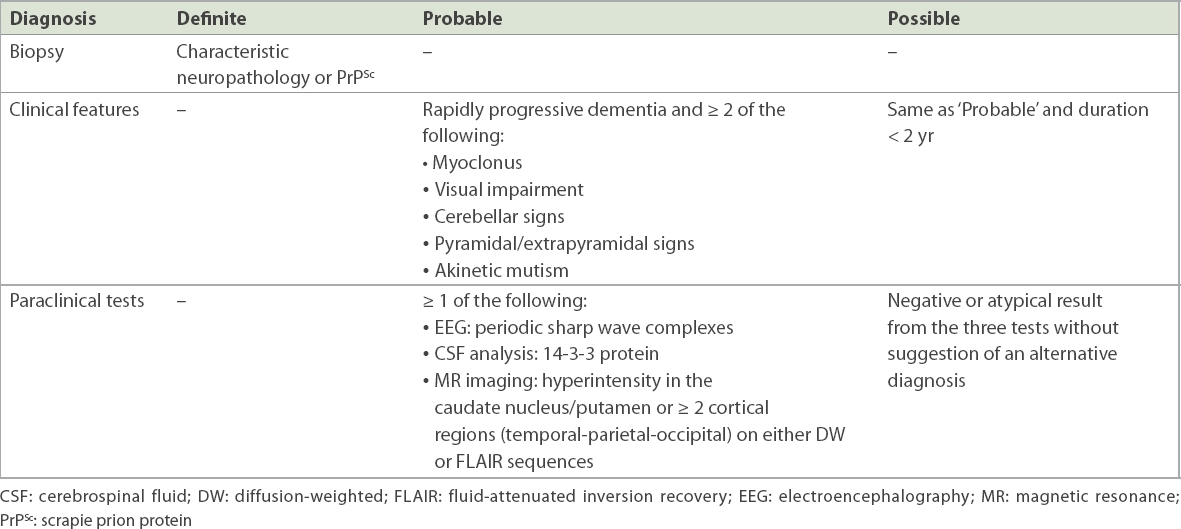
The sine qua non of sCJD is rapidly progressive dementia and invariably fatal outcome; despite this, clinical symptoms can range from isolated visual disturbance, confusion and amnesia to ataxia, dementia and myoclonus.(3) Early diagnosis of sCJD can be challenging due to the variegated signs and symptoms. To facilitate diagnosis, paraclinical tools, such as EEG, and CSF analysis can be used. EEG may show a distinctive pattern of periodic sharp wave complexes, and CSF assays can show the presence of a 14-3-3 protein.(3) MR findings, namely high signal abnormalities in the caudate nucleus and putamen or at least two cortical regions on either DW or FLAIR sequences, have been included in the updated diagnostic criteria of sCJD.(4) These are summarised in
Table I
World Health Organization diagnostic criteria for sporadic Creutzfeldt-Jakob disease.
Neuroimaging, in particular the DW sequence, plays an integral role in supporting the in vivo diagnosis of CJD at an early stage, due to its high sensitivity and specificity. It can give further credence to the reliability of the EEG and CSF results and, in some cases, pre-date abnormal EEG and CSF findings.(6) An abnormal MR image with a typical CJD pattern not uncommonly reveals an unsuspected case of CJD when the EEG and CSF findings are equivocal. Although the pathophysiology of disease manifestation on the DW sequence remains unclear, it has been postulated that restricted diffusion within the affected areas of the brain is related to underlying vacuolation or deposition of the prion protein.(7) Three classic MR imaging patterns of sCJD on DW imaging have been reported: asymmetric involvement of the cortex and basal ganglia; involvement of the cortex only; and involvement of basal ganglia and thalami only.(2,8) Our case demonstrates the most common pattern of CJD, involving both the cortex and basal ganglia. Although the ‘pulvinar’ or ‘double hockey stick’ signs of thalamic involvement are known to be classic radiological hallmarks attributable to variant CJD, they have also been observed in certain molecular subtypes of sCJD.(2) In addition, even when cerebellar signs may be clinically present, MR signal abnormality in the cerebellum is rarely detectable in sCJD. Cerebellar atrophy with elevated diffusivity on apparent diffusion coefficient maps may be the only radiologic sign of cerebellar involvement.(2) As the disease progresses, there may be an increase in both the extent and intensity of DW and FLAIR signal abnormality in the cortex and basal ganglia due to progressive spongiform degeneration. Pseudonormalisation of signal intensity is frequently observed when there is inevitable atrophy in the late stage of the disease process.(6)
Single-photon emission computed tomography and positron emission tomography/computed tomography (PET/CT) are alternative advanced neuroimaging techniques for diagnosing sCJD. PET/CT, in particular 18F-FDG-PET/CT (18F-fluoro-2-deoxy-D-glucose-PET/CT), has proven useful because of its capability of detecting regions of brain tissue with altered metabolic status, which can pre-date abnormalities seen on conventional MR images.(8) This promising imaging tool has not yet been universally utilised due to its lack of availability and accessibility, and therefore currently remains an effective supplement to MR imaging.
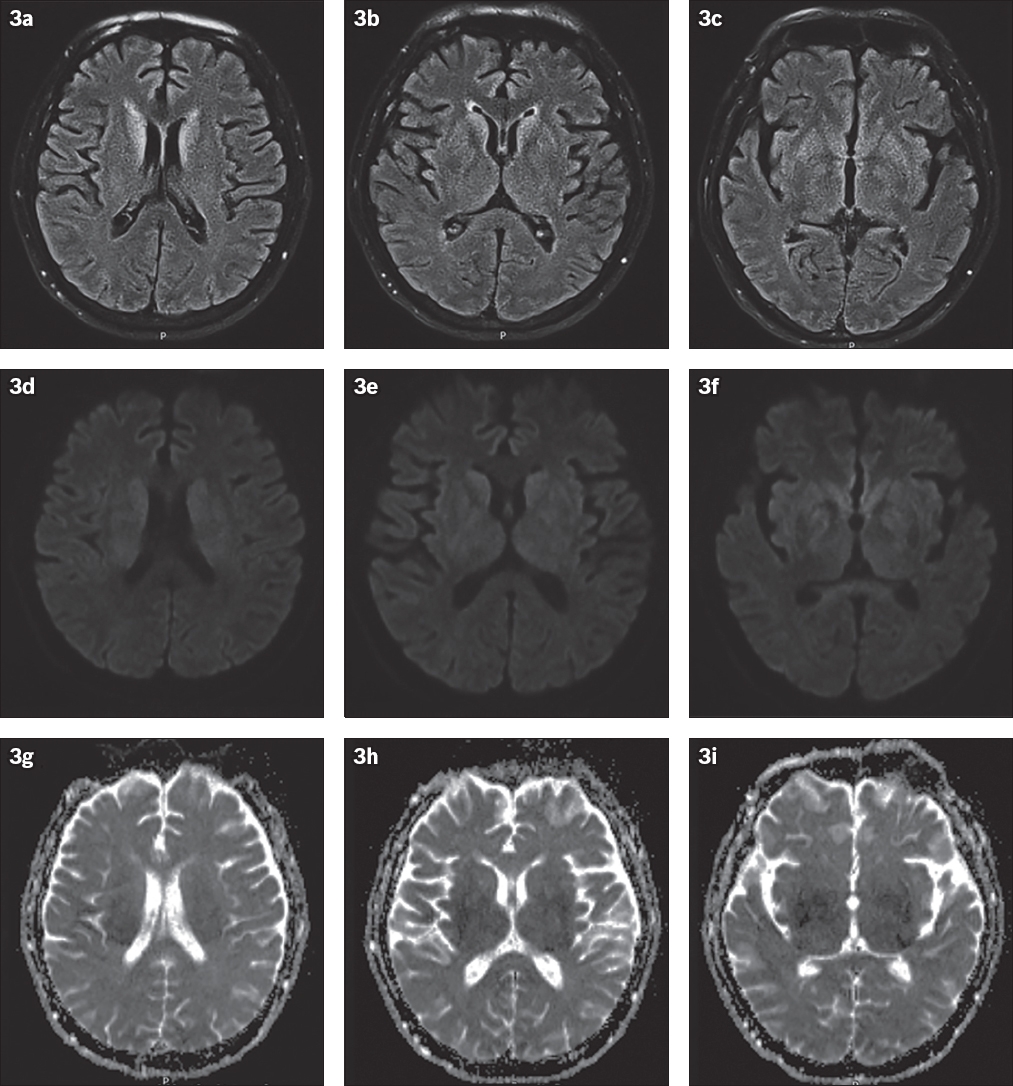
It is important to differentiate sCJD from other neurological diseases with similar clinical or radiologic patterns, because some of the latter may be treatable. We herein introduce some conditions that may mimic sCJD. In the acute setting, hypoxic-ischaemic encephalopathy (HIE) may demonstrate increased signal intensity in the more metabolically active regions of the brain, such as the basal ganglia, hippocampus and cerebral cortex (
Fig. 3
Hypoxic-ischaemic encephalopathy in a 60-year-old man who presented with acute pulmonary embolism. Post-embolectomy, he developed a poor Glasgow Coma Scale score and was placed on extracorporeal membrane oxygenation. (a–c) FLAIR images show symmetrical hyperintensity in the bilateral caudate and inferior lentiform nuclei. (d–f) DW images and (g–i) apparent diffusion coefficient (ADC) map show mildly reduced diffusion in the affected areas.
Herpes encephalitis is the most common acute fatal sporadic viral encephalitis caused by the herpes simplex virus. It can clinically manifest with an acutely decreased level of consciousness, neurological deficits, and nonspecific symptoms such as headache and fever. Unlike sCJD, herpes encephalitis (
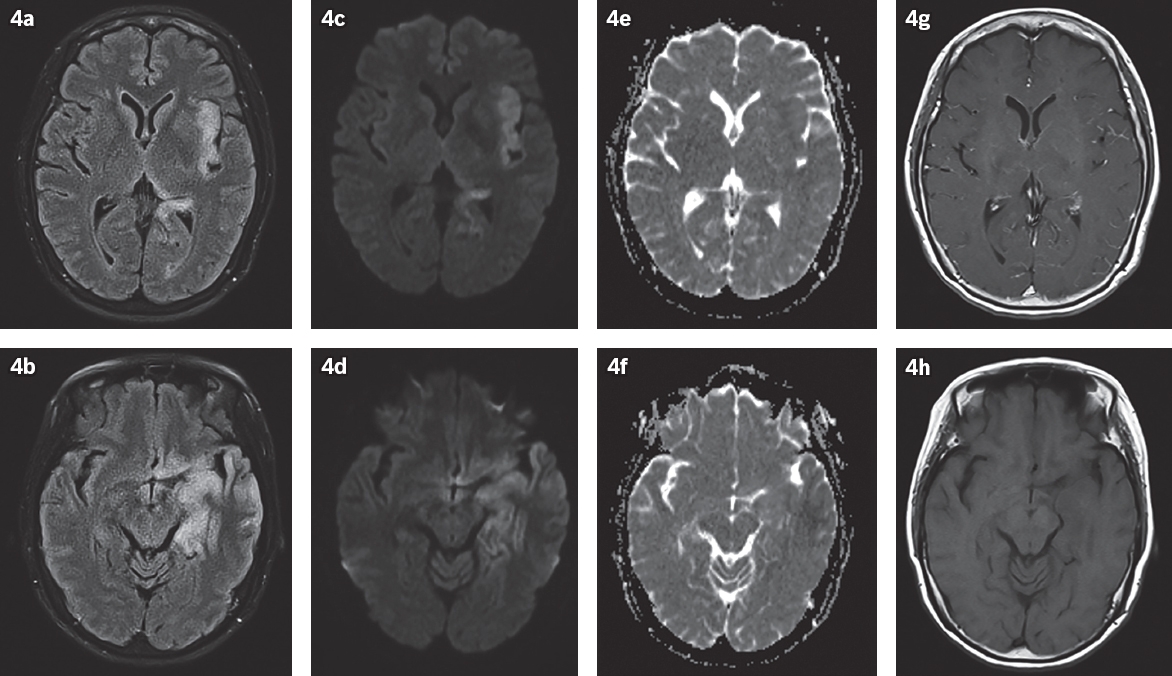
Fig. 4
Herpes simplex virus in a 66-year-old woman who presented with altered mental status, dysphagia, facial asymmetry and high fever. (a & b) FLAIR and (c & d) DW images show abnormal high signal and swelling in the left hippocampus, left mesial and inferior temporal lobe, left insular cortex and left basifrontal lobe, with no (e & f) appreciable corresponding restricted diffusion in these areas on ADC map. (g & h) Post-gadolinium T1-W images show slight leptomeningeal enhancement on the left.
Posterior reversible encephalopathy syndrome is a neurotoxic state resulting from an acute change of perfusional status to the posterior portions of the brain, either due to failed autoregulation or vasoconstriction.(12) The clinical spectrum may range from acute or subacute nonspecific symptoms of headache and altered mental status to visual disturbance and generalised seizure. Vasogenic oedema is classically seen in the posterior parietal and occipital regions, as well as the watershed regions between vascular territories (
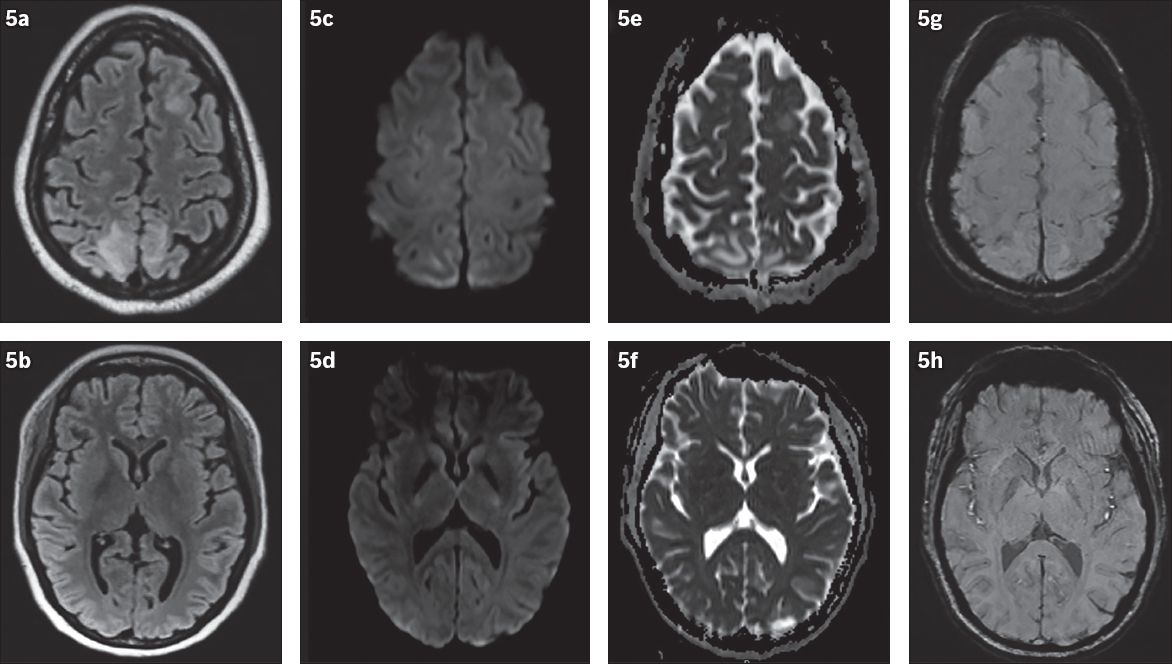
Fig. 5
Posterior reversible encephalopathy syndrome in a 26-year-old woman who presented with seizures and neurodeficits. MR images of the brain show bilateral fairly symmetrical signal abnormalities in the left frontal and bilateral parietal lobes in the cortical and subcortical regions on (a & b) FLAIR and (c & d) DW images, with (e & f) no apparent restricted diffusion in the affected areas and (g & h) no abnormal susceptibility suggesting intracranial haemorrhage. Normal appearance of the basal ganglia is seen in (b).
A postictal state may manifest as transient focal T2/FLAIR and DW signal changes secondary to cytotoxic and vasogenic oedema. Involvement of the hippocampal regions (
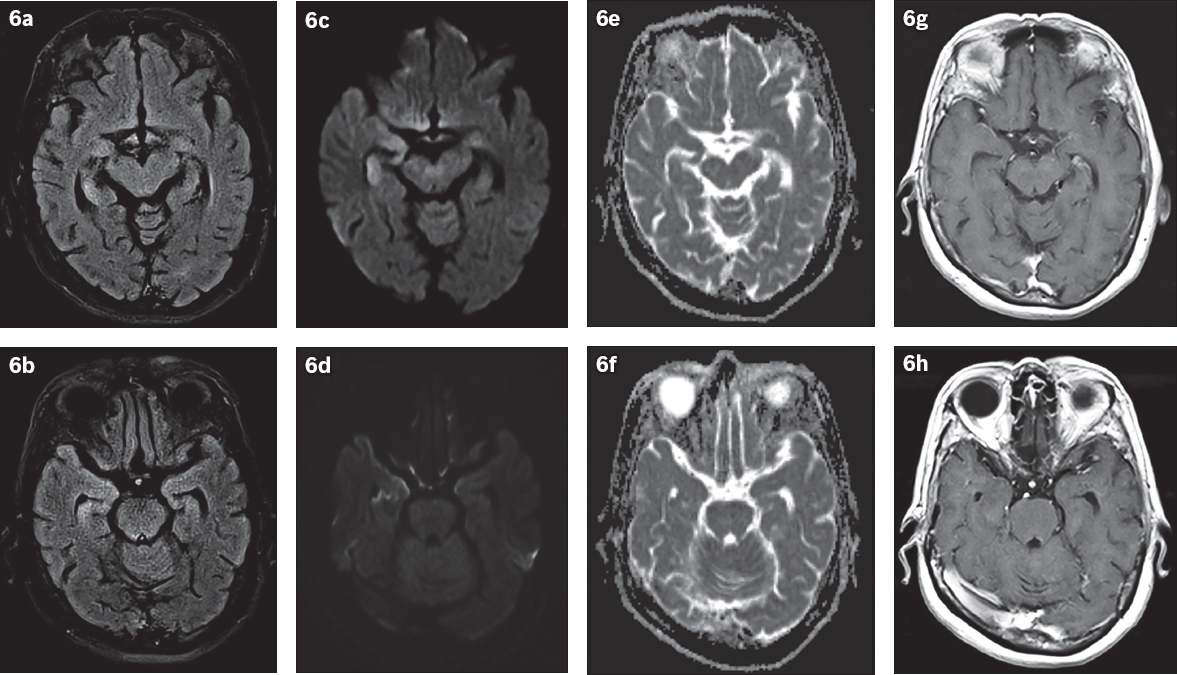
Fig. 6
Postictal state in a 67-year-old woman with a known history of gastrointestinal stromal tumours on imatinib, who presented with seizure. (a & b) FLAIR and (c & d) DW images show increased signal intensity and swelling in the body and tail of the right hippocampus. (e & f) ADC map shows no restricted diffusion detected. The left hippocampus appears unremarkable. (g & h) Post-gadolinium T1-W images show no appreciable abnormal parenchymal or leptomeningeal enhancement.
Voltage-gated potassium channel encephalitis is a common subtype of autoimmune encephalitis defined by intractable epilepsy. The typical MR findings are T2 and FLAIR high signal abnormalities that almost invariably involve the limbic system; however, the cortex and cerebellum may also be involved (
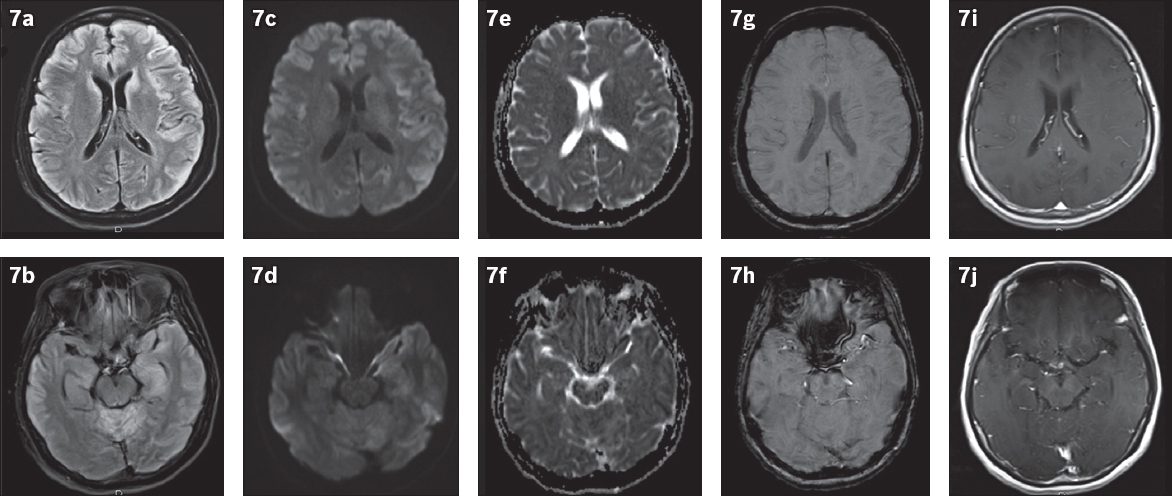
Fig. 7
Voltage-gated potassium channel antibody disease in a 52-year-old woman with suspected encephalitis, who presented with lower limb incoordination and progressive confusion. MR images show (a & b) areas of FLAIR hyperintensity in the bilateral cortices, left hippocampus, bilateral caudate nuclei and cerebellar hemispheres, with (c–f) associated mild diffusion restriction but without (g & h) abnormal susceptibility or (i & j) enhancement.
Wilson’s disease is an acquired hepatolenticular degeneration due to a disorder of copper metabolism and resultant abnormal accumulation of copper in the liver, brain and eyes. The classic MR features are hyperintensity in the basal ganglia, ventrolateral thalamus, midbrain and pons on T2/FLAIR images (
Fig. 8
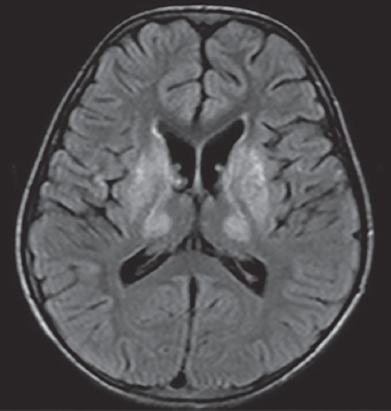
Wilson’s disease in a 15-year-old boy with dysarthria, dystonia and an abnormal liver function test. MR image of the brain shows symmetric abnormal FLAIR hyperintensity in the bilateral basal ganglia and thalami. No cortical signal abnormality is noted.
Wernicke’s encephalopathy is an acute neurological disorder resulting from thiamine (vitamin B1) deficiency, often characterised by the classic triad of oculomotor symptoms, ataxia and altered mental status. This triad can be seen in both alcoholic and non-alcoholic Wernicke’s subtypes. MR imaging typically shows bilateral symmetric signal alterations in the medial thalami, periventricular regions of the third ventricle, mammillary bodies, tectal plate and periaqueductal area (
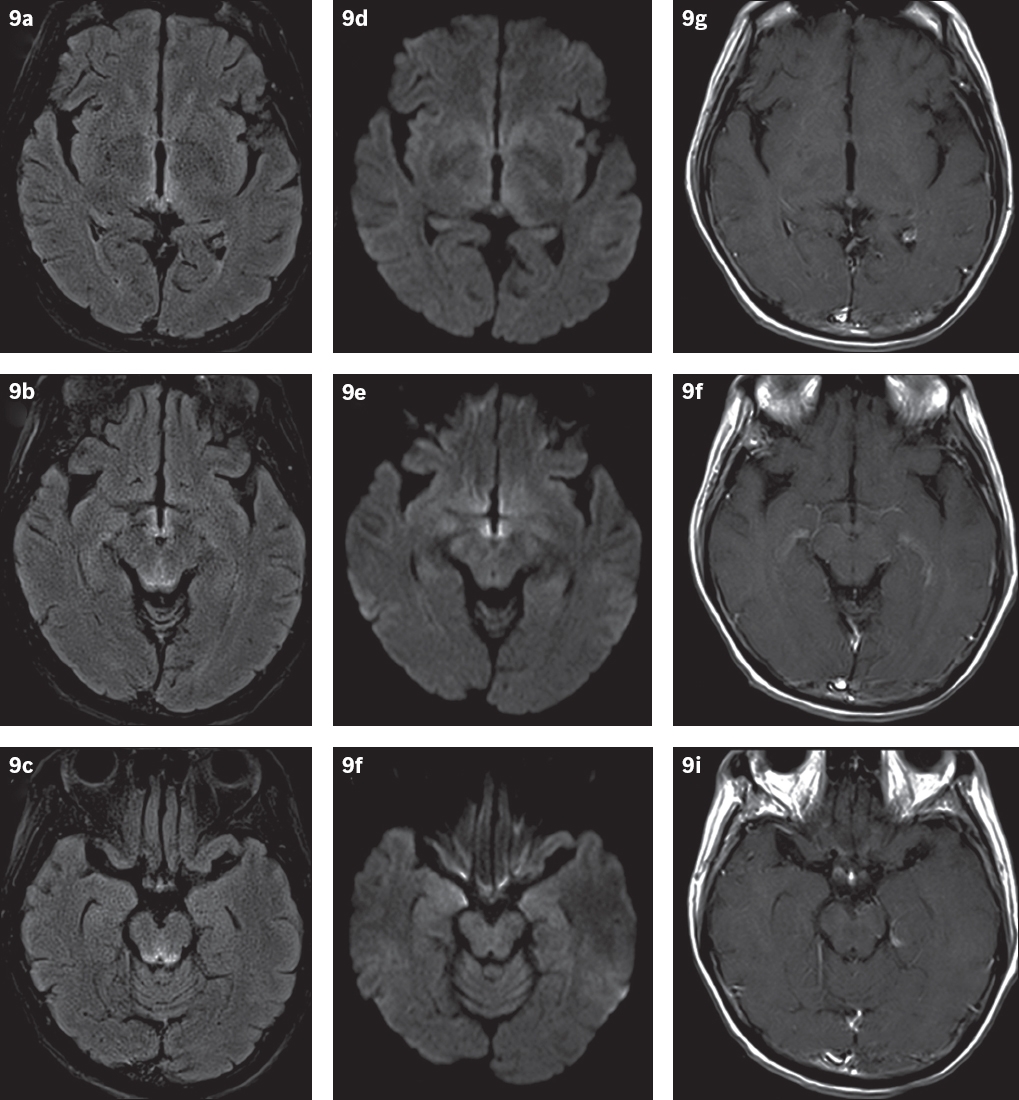
Fig. 9
Probable Wernicke’s encephalopathy in a 59-year-old non-alcoholic female patient with gait disturbance, cognitive impairment and memory loss. (a–c) FLAIR and (d–f) DW images of the brain reveal symmetric hyperintensities in the periventricular region of the third ventricle, medial thalami, tectal plate and periaqueductal region, with no (g–i) associated enhancement. The lack of a history of chronic alcohol abuse suggests that this is a non-alcoholic subtype of Wernicke’s encephalopathy.
In conclusion, based on clinical features alone, it may be challenging to diagnose sCJD in vivo because of its wide spectra of clinical presentations. However, paraclinical biomarkers, and DW imaging in particular, play an integral part in the diagnostic workup of sCJD. Careful interpretation of MR images and recognition of the classic features of sCJD not only facilitate a probable diagnosis but also aid in excluding other mimicking disorders.
SMJ-59-641.pdf