

CASE PRESENTATION
A 75-year-old woman presented in 2014 with a non-resolving left lower lobe consolidation. Initial less invasive attempts at biopsy of the left lower lobe consolidation failed to obtain a definitive diagnosis, and the patient underwent a left lower lobectomy and lymph node dissection. Histology revealed adenocarcinoma with no lymphovascular invasion, and she was diagnosed with Stage IIA lung adenocarcinoma. She did not require radiotherapy and declined adjuvant chemotherapy.
Upper lung changes on initial preoperative computed tomography (CT) of the thorax performed in 2014 (
What do the initial CT done in 2014 (
Fig. 1
Initial CT image of the thorax obtained in 2014.
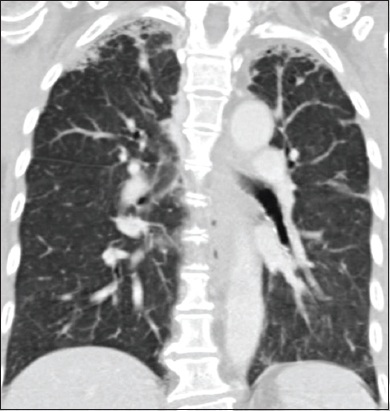
Fig. 2
Subsequent (a) coronal and (b) axial CT images of the thorax obtained in 2019.
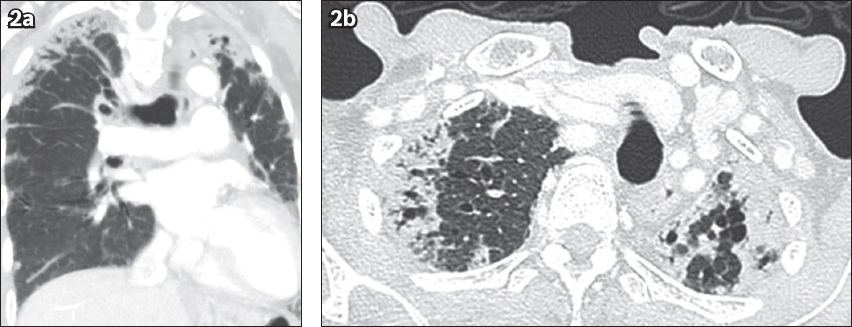
IMAGE INTERPRETATION
Coronal CT of the thorax performed in 2014 (
Follow-up CT of the thorax performed in 2019 (
DIAGNOSIS
Pleuroparenchymal fibroelastosis (PPFE).
CLINICAL COURSE
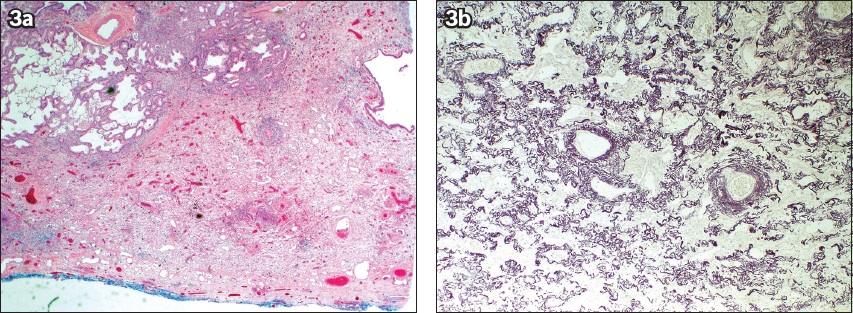
The patient was referred to the interstitial lung disease (ILD) clinic and the case was discussed at the ILD multidisciplinary team meeting. Histology from the left lower lobectomy demonstrated thickening of the pleura as the result of a fibrosing process that was elastotic in nature (Figs.
Fig. 3
(a) Photomicrograph in low-power view shows thickened pleura surface in the resected specimen (Haematoxylin & eosin, original magnification × 40). (b) Photomicrograph in medium-power view shows abundant elastic fibres in the thickened portion of the pleura (elastic van Gieson, original magnification × 100).
DISCUSSION
PPFE is a fibrosing process that affects the pleura and the adjacent lung parenchyma, with a predilection for the upper lobes. Since the initial reports by Amitani et al in 1992,(1) several other corroborative case series have been added through the years, describing its unique clinical, radiological and pathological features that are distinct from other idiopathic interstitial pneumonias (IIPs). This culminated in the recognition of idiopathic PPFE (IPPFE) as a rare IIP in the 2013 joint American Thoracic Society/European Respiratory Society IIP classification.(2)
Although PPFE is classified as a rare IIP, owing to greater awareness since the publication of the IIP classification guidelines and proposal of various diagnostic criteria,(3,4) the incidence and prevalence of PPFE are not as uncommon as once thought. For instance, PPFE accounts for 7.7% of consecutive IIP cases evaluated in a single centre over a ten-year period.(5) The exact pathogenesis of PPFE is poorly understood, although acute and sub-acute lung injury have been proposed to be triggers for this disease. Common aetiologies associated with PPFE include haematopoietic stem cell, bone marrow and lung transplantation.(6) Other associations include allergens and exposure to occupational dust (asbestos, aluminium), connective tissue diseases (scleroderma, rheumatoid arthritis) or exposure to drugs (chemotherapeutic agents, dapsone and daptomycin).(3,7-9) Familial associations have also been reported.(3) Of note, over half of the patients had a history of recurrent pulmonary infections.(3) In the absence of any known associations, the term IPPFE is used.
PPFE affects patients of a wide age range. The median age at presentation is in the fifth decade of life, and the age distribution is likely to be bimodal, peaking at the third and sixth decade.(6,10) It does not have any gender preponderance. Common presenting symptoms include insidious onset of progressive dyspnoea, cough and weight loss. With progression of PPFE or when it co-exists with other fibrotic lung diseases, lung auscultation may reveal inspiratory crackles. One peculiar sign of PPFE is the development of platythorax, also called the ‘flattened thoracic cage’ appearance, due to upper lobe contraction and reduced chest wall bulk.(7,11)
Imaging is often the first step in the diagnosis of PPFE. In the early stages, chest radiography shows bilateral upper zone pleural thickening, which may sometimes be asymmetrical. With progression of fibrosis, there is lobar volume loss, often accompanied by hilar elevation.(12) On high-resolution CT images of the chest, Reddy et al defined ‘definite PPFE’ as upper lobe pleural thickening with associated subpleural fibrosis, with absent or minimal involvement of the lower lobes.(3) The fibrosis is manifested as architectural distortion and traction bronchiectasis. Other ancillary CT findings include anteroposterior flattening of the chest (platythorax), ‘free-standing’ bronchiectasis and mosaic attenuation of the lung parenchyma.(7,11) Other patterns of lung fibrosis can co-exist with PPFE, most frequently usual interstitial pneumonia, non-specific interstitial pneumonia or hypersensitivity pneumonitis.(7) Figs.
Fig. 4
A 79-year-old man had pleuroparenchymal fibroelastosis with co-existent idiopathic pulmonary fibrosis. Coronal CT image shows bilateral upper lobe subpleural fibrosis and wedge-shaped subpleural consolidation, with honeycombing at the lung bases. Spirometry showed a forced vital capacity of 37% predicted and raised residual volume/total lung capacity ratio of 128%. The body mass index of the patient was 14.6 kg/m2. He died two months after his diagnosis.
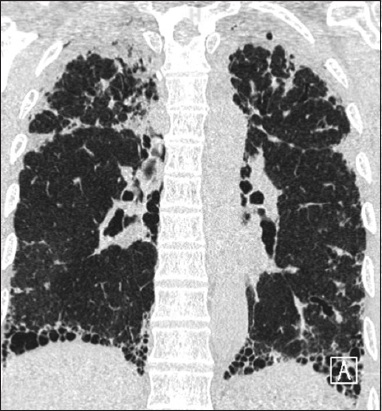
Fig. 5
CT images of a 71-year-old man with pleuroparenchymal fibroelastosis show (a) bilateral subpleural fibrotic changes with traction bronchiectasis in the upper lobes; (b) ‘free-standing’ bronchiectasis, which may reflect recurrent aspiration, in the middle lobe; and (c) a typical interstitial pneumonia pattern with subpleural reticulation, traction bronchiectasis and honeycombing in the lung bases.
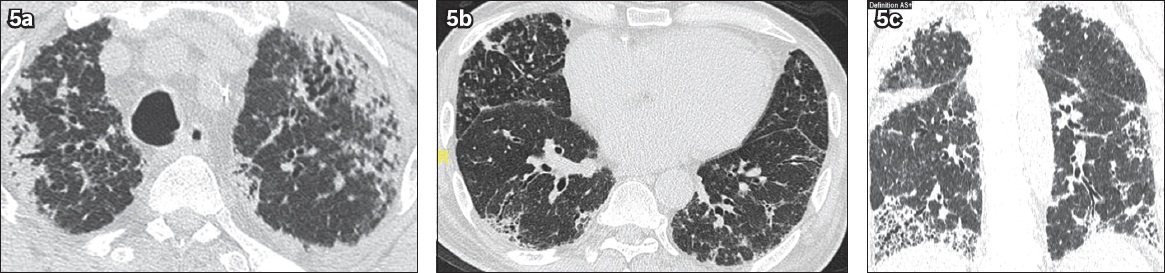
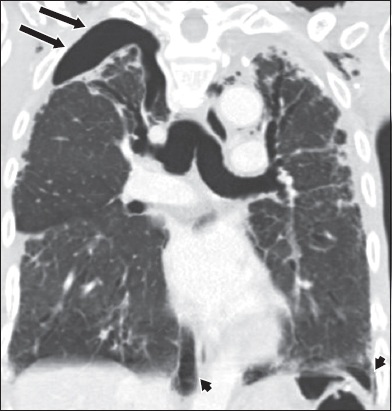
Fig. 6
Pleuroparenchymal fibroelastosis was observed in a 63-year-old man who presented with a five-day history of dyspnoea. Coronal CT image of the thorax shows a right apical pneumothorax (arrows) and at both lung bases (arrowheads).
Several differential diagnoses should be considered when evaluating patients with upper zone fibrosis. Benign pathologies such as apical pleural caps are often incidental findings on imaging in asymptomatic elderly smokers. Unlike PPFE, they are typically localised at the uppermost 5 mm of the lung apices. Differentiating PPFE from active pulmonary tuberculosis (TB) is usually straightforward, as the latter presents with asymmetrical upper lobe centrilobular nodules, consolidation or cavities. However, differentiating PPFE from sequelae of past tuberculous pleurisy is more challenging, as the resultant pleural thickening and calcification from TB may mimic the findings of PPFE (
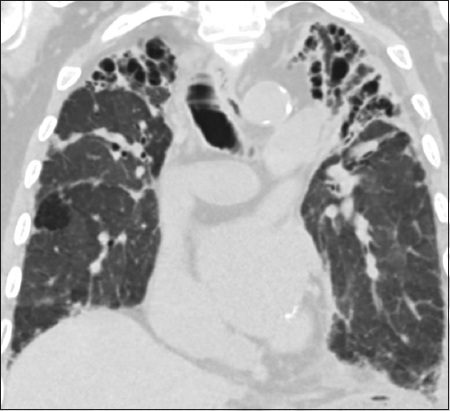
Fig. 7
An 82-year-old man had a history of pulmonary tuberculosis diagnosed and treated more than 30 years ago. Coronal CT image of the thorax shows bilateral, symmetrical upper lobe fibrosis, volume loss with associated severe traction bronchiectasis and biapical pleural thickening. Sequelae of prior tuberculosis is a common mimic of pleuroparenchymal fibroelastosis in the local context and can pose a challenge in diagnosis.
A surgical lung biopsy is required to secure the diagnosis of PPFE. Histologically, PPFE demonstrates intense visceral pleural fibrosis, and prominent and homogenous subpleural intra-alveolar fibrosis with alveolar septal elastosis that is best visualised with an elastin van Gieson stain. The alveolar septal elastosis spares the lung parenchyma away from the pleura, with scanty lymphoplasmacytic infiltrates and small numbers of fibroblastic foci at most.(12) There could be some degree of temporal continuity from early interstitial inflammation and fibrosis to the eventual development of PPFE as well as the possibility of co-existent ILDs.(13)
As surgical lung biopsies may result in postoperative complications such as pneumothoraces and prolonged air leaks, IPPFE diagnostic criteria without the need for histology were proposed recently by Watanabe et al. Using imaging, clinical and physiological features, patients were labelled as ‘radiologically possible IPPFE’, ‘radiologically probable IPPFE’, or ‘radiologically and physiologically probable IPPFE’.(4) ‘Radiologically possible IPPFE’ included early imaging features of upper lobe subpleural airspace consolidation with traction bronchiectasis, while ‘radiologically probable IPPFE’ required additional progressive features of upper lobe volume loss or bilateral upward shift of hilar structures to be present, together with clinical features such as dry cough or exertional dyspnoea. ‘Radiologically and physiologically probable IPPFE’ further included physiological parameters such as the percentage predicted value of the residual volume/total lung capacity ratio as well as the BMI of the patient. However, none of these classifications have been validated in other studies.
No treatment has been shown to be effective in PPFE, except for lung transplantation. The long-term prognosis of PPFE varies from an insidious course over 10–20 years to a more progressive respiratory decline, with a median survival of less than five years.(7) Transplant-associated PPFE may portend a poorer prognosis,(6) and patients with PPFE co-existing with idiopathic pulmonary fibrosis have more frequent complications and poorer survival.(14)
This case highlights several learning points. Firstly, given the various associations with PPFE, obtaining a detailed history is important when evaluating a patient presenting with radiological features suggestive of PPFE. As TB is prevalent in this region, eliciting symptoms such as chronic cough, haemoptysis and constitutional symptoms, or a previous history of TB may prompt the clinician to obtain sputum for microbiological correlation. Secondly, even though PPFE is described as predominantly affecting the upper lobes, it can also involve the lower lobes, although the changes may be more subtle. Finally, it is important to relook and consider alternative diagnoses, especially when the patient’s clinical behaviour is not in keeping with expected disease trajectory, as demonstrated by this case.
In conclusion, greater awareness of PPFE has enabled more in-depth understanding of its pathogenesis, temporal evolution with time and longitudinal disease course. Establishing universally accepted diagnostic criteria may help to facilitate studies in a more uniform cohort to better understand this disease.
Supplementary Material
SMJ-63-78.pdf